
Abstract
Abstract
Schizophrenia, a complex neurological disorder, is comprised of interactions between multiple genetic and environmental factors wherein each of the factors individually exhibits a small effect. In this regard a network-based strategy is best suited to capture the combined effect of multiple genes with their definite pattern of interactions. Given that schizophrenia affects multiple regions of the brain, we postulated that instead of any single specific tissue, a mutual set of interactions occurs between different regions of brain in a well-defined pattern responsible for the disease phenotype. To validate, we constructed and compared tissue specific co-expression networks of schizophrenia candidate genes in twenty diverse brain tissues. As predicted, we observed a common interaction network of certain genes in all the studied brain tissues. We examined fundamental network topologies of the common network to sequester essential common candidates for schizophrenia. We also performed a gene set analysis to identify the essential biological pathways enriched by the common candidates in the network. Finally, the candidate drug targets were prioritized and scored against known available schizophrenic drugs by molecular docking studies. We distinctively identified protein kinases as the top candidates in the network that can serve as probable drug targets for the disease. Conclusively, we propose that a comprehensive study of the connectivity amongst the disease genes themselves may turn out to be more informative to understand schizophrenia disease etiology and the underlying complexity.
Introduction
S
Recent network-based studies done in schizophrenia and other neurological complex disorders focus on a protein–protein interaction network (PPIN) where a network is constructed using disease genes and their interacting partners or nearest neighbors from the human interactome (Camargo et al., 2007; Lee et al., 2011; Luo et al., 2014). Diverse approaches are being used to select candidate genes for network construction. The use of differentially expressed and co-expressed gene information is one of the popular strategies for network generation (Hsu et al., 2008; Sreedevi and Danail, 2012; Torkamani et al., 2010). Undoubtedly, these expression networks reveal interaction among important genes, but the approach is constrained by data normalization and false discovery rate (FDR) correction methods. Similarly, mapping GWAS identified disease-associated variants in the schizophrenia risk gene network highlights crucial nodes in the network (Gilman et al., 2012; Jia et al., 2012; Yu et al., 2014). However, the population sample size and stringent statistical parameters: p-value distribution, multiple testing correction methods, etc. warrant the utility of the method. Another method used is a gene prioritization approach, where candidate genes reported from genetic studies such as linkage, association, and gene expression are scored and used to structure a molecular gene network (Sun et al., 2009). However, the prioritization approach is also limited by accuracy of the scoring method. Recently, gene regulatory networks are also being employed to understand the disease complexity (Guo et al., 2010; Potkin et al., 2010). Regulatory networks are certainly meaningful in understanding the regulation of disease genes; still the strategy is constrained by valid experimental proof for regulation of disease genes.
Several histochemical studies reveal that schizophrenia primarily affects two core areas of the cerebral cortex: the parietal lobe and the frontal lobe (Huang et al., 2009; Premkumar et al., 2006; Tu et al., 2013; Yildiz et al., 2011). Schizophrenic patients exhibit significantly reduced expression of glutaminergic and dopaminergic receptors in the frontal lobe (Carlsson et al., 1990; Tseng et al., 2007), while the parietal lobe undergoes significant decrease in gray matter volume (Zhou et al., 2007). Also, corpus callosum (the white matter connecting the two cerebral hemispheres) changes in shape and size (Woodruff et al., 1995). Apart from this, reduction in the volume of temporal lobe has been associated with positive symptoms of schizophrenia (Andreasen et al., 1990; Barta et al., 1990, and the thalamus reduces in size leading to hypometabolism in schizophrenia (Andreasen et al., 1994).
In view of involvement of different regions of the brain, we hypothesize that instead of any individual specific tissue, a mutual set of interactions operate between these regions in a defined pattern that lead to a disease phenotype. To validate, we constructed a tissue specific co-expression network of schizophrenia candidate genes for various parts of the brain. As predicted, we found a set of interacting candidate pairs that are common in each tissue, suggestive of a shared interaction network for schizophrenia in brain. To support the above finding, we also studied the network topology of nodal genes in the common network in contrast to tissue-specific networks so as to identify probable drug targets in schizophrenia. In conclusion, we suggest that the disease genes operate via a distinct pattern while interacting with each other in different biological pathways. Understanding interactions amongst these candidate disease genes will be more meaningful to ascertain the underlying complexity of this disease.
Methods
Source of the dataset
Schizophrenia disease genes (SDG) were retrieved from four well-characterized public databases. The initial dataset was extracted from the OMIM MorbidMap (Amberger et al., 2009) consisting of 63 SDGs. The second dataset was derived from DisGeNet database (Bauer-Mehren et al., 2011), a comprehensive resource of gene-disease association that encompasses biomedical aspects of many diseases and provides vast information of disease genes. We found 553 SDG on searching the database with the keyword ‘schizophrenia.’ The third and fourth datasets were from NeuroDNet (Vasaikar et al., 2013) and Schizophrenia Gene Resource (SZGR) (Jia et al., 2010), two well-known resources for human neurological disorders and associated genes. A total number of 25 and 38 candidates SDG were found in these two databases, respectively. The compilation of SDG from all four datasets and on removal of redundant genes resulted in 556 unique disease genes for schizophrenia. Ethical approvals were not considered as the datasets were retrieved from publically available databases and no clinical samples have been used in this study.
Construction of disease gene interaction network
We constructed a schizophrenia disease gene co-expression network using a modified version of CRG interactome (Bossi and Lehner, 2009). CRG interactome is by far the largest protein–protein interaction network (PPIN) that integrates data from 21 known PPIN database resources to define a network of 80,922 physical interactions that can occur between 10,229 human proteins. Each pair of interactions was further extrapolated with their co-expression information in 78 different human tissues from GNF Atlas database (Su et al., 2004). An interaction is reported for a pair considering a normalized threshold expression value of 200. Any two genes are classified as co-expressed (1) if their normalized expression value is more than 200 and as not co-expressed (0) if the normalized expression is less than 200 (Bossi and Lehner, 2009). To confirm proper annotation, we retrieved Ensembl gene identifiers for 556 SDG and mapped them to CRG interactome. Resultant interactions were plotted as an undirected graph where each node is represented by the SDG and the two nodes are connected by an edge, if their partners are co-expressed in at least one tissue type in CRG data. We used an open source software platform, Cytoscape; v2.8.7 (Shannon et al., 2003) for visualization and further analysis of the network. The importance of a node in a molecular network is often correlated to its centrality (Jeong et al., 2001). There are different measures that capture the centrality of a node in a network. We investigated three different centrality measures namely Degree, Betweenness, and Closeness to prioritize the importance of SDG in the network. Details of these parameters have already been discussed in our earlier study (Podder et al., 2014).
All the measurements describing the network topology were calculated using NetworkAnalyzer (Assenov et al., 2008) which is a java based application (plugin) for Cytoscape.
Classification of housekeeping and non-housekeeping genes
Housekeeping (HK) genes are universally expressed in all tissues and their constitutive expression is essential to maintain the cellular functions (Butte et al., 2001). The expression levels of HK genes have been found to be constant in many diseased tissues as well as in normal tissues (Cheng et al., 2011; Morgan et al., 2010). Therefore, it is necessary to look at HK genes if they contribute fairly in disease pathophysiology. A list of human housekeeping genes (HK) was compiled from two different datasets by Chang et al., (2011) and Eisenberg and Levanon, (2013). In the first study (Chang et al., 2011), a meta-analysis has been performed, where genes from 104 different microarray datasets have been catalogued from 43 normal human tissues that identified 2064 housekeeping genes. On the other hand, in the second study (Eisenberg and Levanon, 2013), an expression profiling of publicly available RNA-seq data identified 3804 genes as HK genes whose uniform expression was observed across a panel of 16 normal human tissues. In the present work, we have considered the HK genes from both these studies. It was observed that less than 50% of HK genes overlapped in both studies. The redundant genes were removed, which resulted in a total of 4694 genes as HK in humans. Schizophrenia disease gene interactions were so classified based upon the presence of housekeeping (HK) disease genes and non-housekeeping (NHK) disease genes in the network.
Identification of candidate pathways
Biological pathways were identified from KEGG database (Kanehisa et al., 2012) using gene set enrichment analysis. We implemented the method described in ClueGo plugin (Bindea et al., 2009) for enrichment calculations. The p-values were calculated by a two-sided hypergeometric test followed by the Bonferroni correction (p≤0.05) for each KEGG term to determine the list of significant pathways (Rivals et al., 2007).
Identification of drug targets and docking studies
DrugBank3.0 (http://www.drugbank.ca/) (Knox et al., 2011) is currently the most comprehensive drug database that facilitates the study of multiple genes and drugs involved in a specific biological process. To identify the drugs related to schizophrenia, we used ‘schizophrenia’ as a search term and retrieved the specific list of drugs. A compilation of schizophrenic drugs together with their targets have been reported in a recent study (Putnam et al., 2011). We finally enlisted 48 drugs along with their targets. Additionally, docking studies were performed for a set of prospective drug targets in the network against all available schizophrenia drugs by using extra-precision (XP) mode of the Glide cross docking program (Schrödinger suite, Maestro version 9.3) (Friesner et al., 2006). Binding free energy (ΔG) for each docked pose was further estimated using MM-GBSA calculations (Prime, version 3.1) (Rastelli et al., 2010).
Results
Tissue-specific Disease Genes Interaction Network (DGNet)
We considered 556 schizophrenia disease genes (SDG) from the current databases to construct the disease gene co-expression network. Schizophrenia being a neurological disorder, we mapped SDG in twenty brain-related tissues in CRG data to compute the key interactions in different parts of human brain. In order to check the co-expression, an interaction pair was selected if both the genes in the SDG list correspond to a co-expression value of 1. We employed an in-house perl script to retrieve such interaction pairs in all the twenty tissues. Further, interaction pairs for each tissue were mapped together to form an undirected co-expression network of disease genes in schizophrenia (DGNet). We constructed twenty such DGNet. The numbers of disease genes and their interaction details for all the twenty tissues are presented in Figure 1. Majority of the interactions were seen in hypothalamus (206 SDG along with 322 interactions) followed by other tissues in brain. We noticed that the number of SDG (187–206) and their interactions (277–322) in each tissue did not differ remarkably, hence indicating that a set of SDG might share common interactions across those tissues.
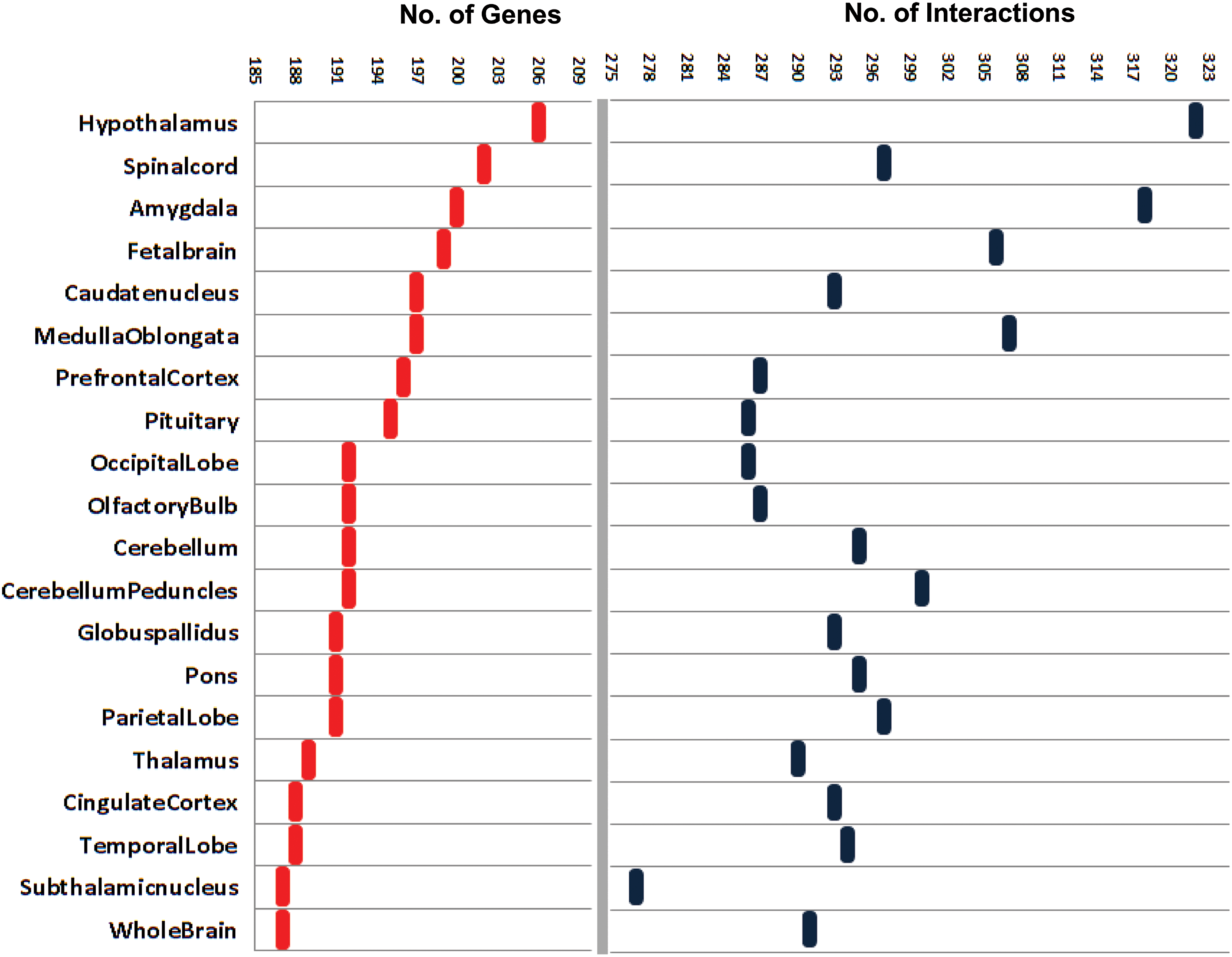
SDG interaction details in twenty different tissues of brain. Tissues are represented in descending order based upon the number of SDG (Schizophrenia Disease Gene) present in each of them followed by their aggregate interaction statistics.
We also calculated the degree distribution (Diestel, 2005) of SDG in each of the twenty networks. The degree is the number of connections or edges that a SDG makes with any other node in the network. A comparison of degree distribution plot within the DGNet revealed that all of them (DGNet) followed the power law distribution that is a scale-free property of any biological network (Barabasi et al., 1999). We also observed that the average number of nodes having similar degree in the network is merely the same (Fig. 2) reflecting a commonality within the DGNet.

Degree distribution of DGNet in twenty different tissues of brain. The degree distribution curve of DGNet follows the power law distribution in each of the twenty tissues of brain. The comparison plot signified, the average number of nodes those shared similar degree across the networks are nearly overlapping with each other. The power curve shows the distribution of DGNet in hypothalamus with R2 value 0.899.
Commonality within DGNet and expansion to (DG+NDG)Net
We evaluated the commonality within the SDG interaction pairs in all twenty tissues by an in-house developed perl script. We identified a total of 155 paired interactions among 113 SDG that were common to all twenty tissues. Based on the above information, a common interaction co-expression network of disease genes in schizophrenia was designed (Fig. 3). To investigate the diverse role of SDG, we further expanded the common DGNet in the human interactome where we included the interaction information of SDG as well as NSDG (Non-Schizophrenia Disease Genes) to structure the common ‘Disease Genes+Non-Disease Genes’ interaction Network (DG+NDG)Net. The mutual interactions between NSDG were ignored in the resultant network to reduce the complexity. Similar networks were also constructed in tissue specific manner for hypothalamus (Supplementary Fig. S1). We finally arrived at four separate but interrelated networks namely the DGNet and (DG+NDG)Net for hypothalamus and the common tissue, respectively, for further investigations. Details about the information on networks have been provided in Supplementary Figure S2. We observed that in (DG+NDG)Net, less than 8% genes belonged to SDG set, and rest of the genes were NDG (not for schizophrenia). Interestingly, a subsequent reduction of the network density from the tissue-specific network (hypothalamus) to the common interacting network of SDG also resulted in the same ratio.
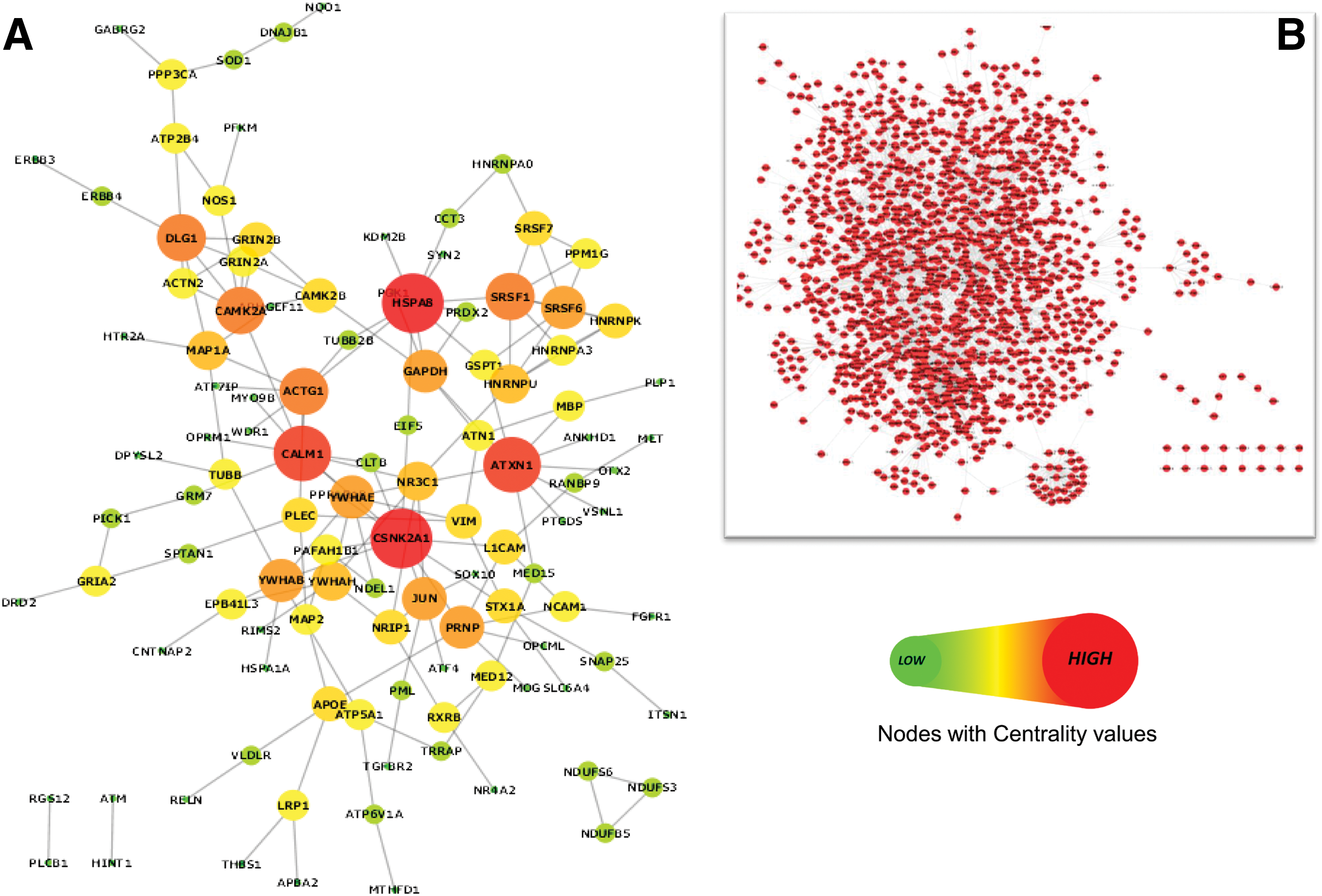
Common interacting (co-expression) network of SDG in brain.
Comparison Between DGNet and (DG+NDG)Net
Network centrality measurement
Basic topological parameters of each network were taken for network centrality measurement. We calculated Node Degree, Betweeness Centrality (BC), and Closeness Centrality (CC) of each node in all four networks and compared the results. BC of a node calculates the number of shortest paths that passes through a particular node in the network (Brandes, 2001), whereas nodes with high CC value have the ability to contact any node in the network in the shortest possible path (Freeman, 1979). Altogether, these parameters evaluate the physical importance of any node in the network. In biological networks, genes with rich topological values mostly tend to work as a central regulatory “hub” (Vázquez et al., 2004). We plotted the BC and CC values together for the corresponding genes of both the DGNet and (DG+NDG)Net present in common interaction network. We found in both the cases that the genes with higher topological (BC and CC) values were mostly shared (Fig. 4). We further compared the degree value of each node between the networks in hypothalamus and the common interacting network of SDG (Table 1). We found certain genes like HSPA8, CSNK2A1, CALM1, ATXN1, SRSF1/6, YWHAB, and JUN were retained as highly connected nodes in all four networks. On decreasing the network density, the number of interactions made by each node altered without affecting the priority of nodes. Interestingly, we identified few known candidate genes for schizophrenia such as DISC1 and YWHAG as important members in hypothalamus network that lacked in the common interacting network of SDG.
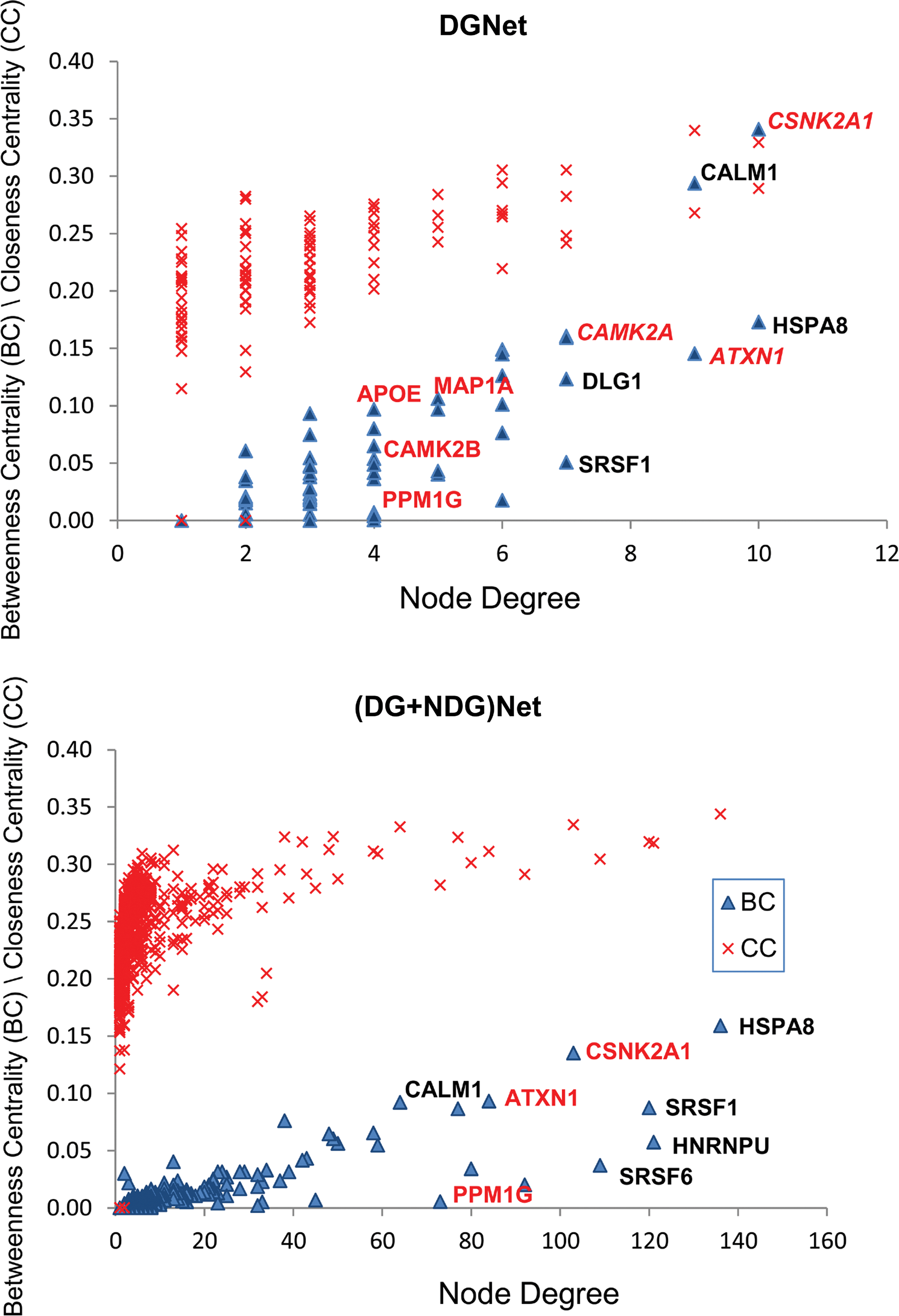
Comparison of topological parameters between DGNet and (DG+NDG)Net. The Betweenness centrality (BC) plot (green triangle at the bottom) and the Closeness centrality (CC) plot (red cross at the top) in each graph shows the distribution of nodes in corresponding network. Topologically enriched nodes became common and highlighted in both networks.
A comparison of node degree values between DGNet and (DG+NDG)Net in both hypothalamus and common interacting network of SDG. Degree distribution of top 25 genes in each network reveal that essential SDGs appear highly connected in all four different networks (highlighted in
Presence of Housekeeping and Non-housekeeping Genes
To further investigate, we classified the DGNet and (DG+NDG)Net of common interacting network based on the presence of housekeeping (HK) and non-housekeeping (NHK) genes. In both cases, the distribution of HK genes clearly reflected that when we incorporate the interactions of NDG in DGNet of SDG the percentage of HK genes gradually increased from 30% to 66% (Fig. 5). On examination of the topologically enriched genes in both the common interacting networks (Fig. 4), we found that the top identified genes like HSPA8, CALM1, DLG1, and genes from SRSF families fall in the HK category, reflecting their universal role to maintain certain biological functions in each and every tissue of the brain. Also, throughout our study, we were able to isolate common NHK gene-rich interactions (CSNK2A1, CAMK2A, CAMK2B, ATXN1, and PPM1G) that were retained in both the common interacting network of brain.
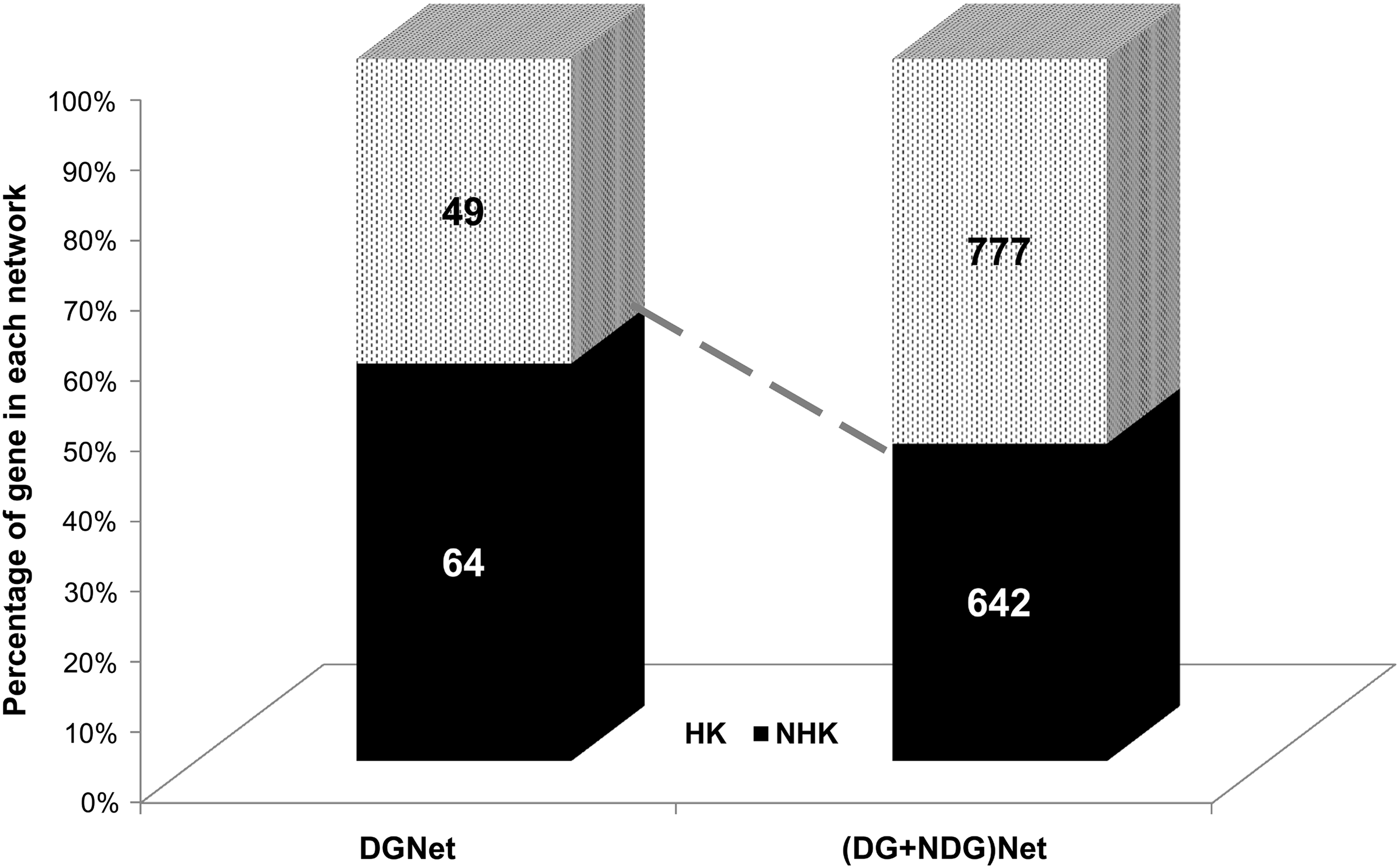
Distribution of HK and NHK genes in DGNet and (DG+NDG)Net. 4694 human housekeeping genes (HK) were compiled from two different datasets (Chang et al., 2011; Eisenberg et al., 2013). Genes present in both the DGNet and (DG+NDG)Net were systematically classified into two separate groups: housekeeping (HK) and non-housekeeping (NHK). An increased ratio of HK genes was observed when the interactions of NDG were included with SDG.
Knowledge of candidate pathways
We next performed gene set enrichment analysis of DGNet present in both hypothalamus and common interacting network of SDG. Significant biological pathways for schizophrenia enriched by SDG in each network were identified from KEGG database (Table 2). The comparison chart clearly highlights important candidate pathways for schizophrenia such as amphetamine addiction (KEGG ID: 5031) (p=4.31E-11 and 1.62E-08) and dopaminergic synapse (KEGG ID: 4728) (p=1.36E-09 and 1.07E-05), respectively, in both the networks. Other essential pathways, such as neuroactive ligand-receptor interaction (KEGG ID: 4080), calcium signaling (KEGG ID: 4020), and neurotropin signaling (KEGG ID: 4722) were also sustained in both the networks throughout the process. The above observation led to the conclusion that identification of common interaction network of disease genes will help in prioritizing significant pathways similar to those obtained in tissue specific network. Hence, we suggest that the common DGNet is robust enough to capture the important pathways in brain that are generally affected in various tissues in schizophrenia. Interestingly, we also observed some significant overlapping gene sets with other disease pathways like Alzheimer's (KEGG ID: 5010) and amyotrophic lateral sclerosis (ALS) (KEGG ID: 5014). The overlapping SDG might act as ‘pleotropic genes’ that are characteristic of disease genes present in phenotypically similar complex disorders (Aerts et al., 2006).
p-value was calculated by two-sided hypergeometric test. Correction of p-value has been done using Bonferroni correction method. P-value less than 0.05 considered significant. The KEGG database search and analysis was performed using ClueGO (Cytoscape plugin).
Prioritization of drugs and drug-targets
To prioritize important drugs and their targets for the DGNet, we designed a drug–target interaction network based on available drug information for schizophrenia from DrugBank (Knox et al., 2011). We identified 48 known schizophrenia-specific drugs along with their targets. Further, each candidate SDG was matched with the targets in the DGNet. Two receptor protein encoded genes from dopamine family (DRD2) and serotonin family (HTR2A) were identified as major drug targets in the common DGNet. Currently, there are 37 and 32 drugs available for both the targets, respectively. Few G-protein encoded genes (GRIN1, GRIN2A, GRIN2B, and GRIA) were found to be targeted by the drug L-glutamic acid (DB00142). Additionally, calcium signaling protein (CALM1) was also identified to be targeted by the known drugs like fluphenazine (DB00623) and trifluoperazine (DB00831).
However, based on the existing databases in the public domain, we were unable to detect any drug for the novel candidate genes that were found to be topologically enriched in the identified common DGNet. Therefore, we employed a molecular docking strategy where the top ranked genes were docked against all available schizophrenia specific drugs from DrugBank. We performed a comprehensive search of all 113 genes of common DGNet in the Protein Data Bank (PDB) (Berman et al., 2000). A total of 23 crystal structures were identified, out of which only 11 were NHK genes. Information on the PDB structures and drug targets is listed in Supplementary Table S1. Top candidates identified from the common DGNet belonged to the protein kinase family (CSNK2A1, CAMK2A, and CAMK2B). Availability of the crystal structures of these protein kinases allowed us to perform molecular docking and followed by calculation of binding affinities with schizophrenia specific drugs (Fig. 6). We found that drugs such as lurasidone (DB08815), aripiprazole (DB01238), and bromocriptine (DB01200) exhibited greater binding affinity for all the three kinases (ΔG>− 80 kcal/mol). A few bridge drugs such as haloperidol (DB00502), trazodone (DB00656), and trifluoperazine (DB00831) also showed significant binding energy (ΔG>− 70 kcal/mol).
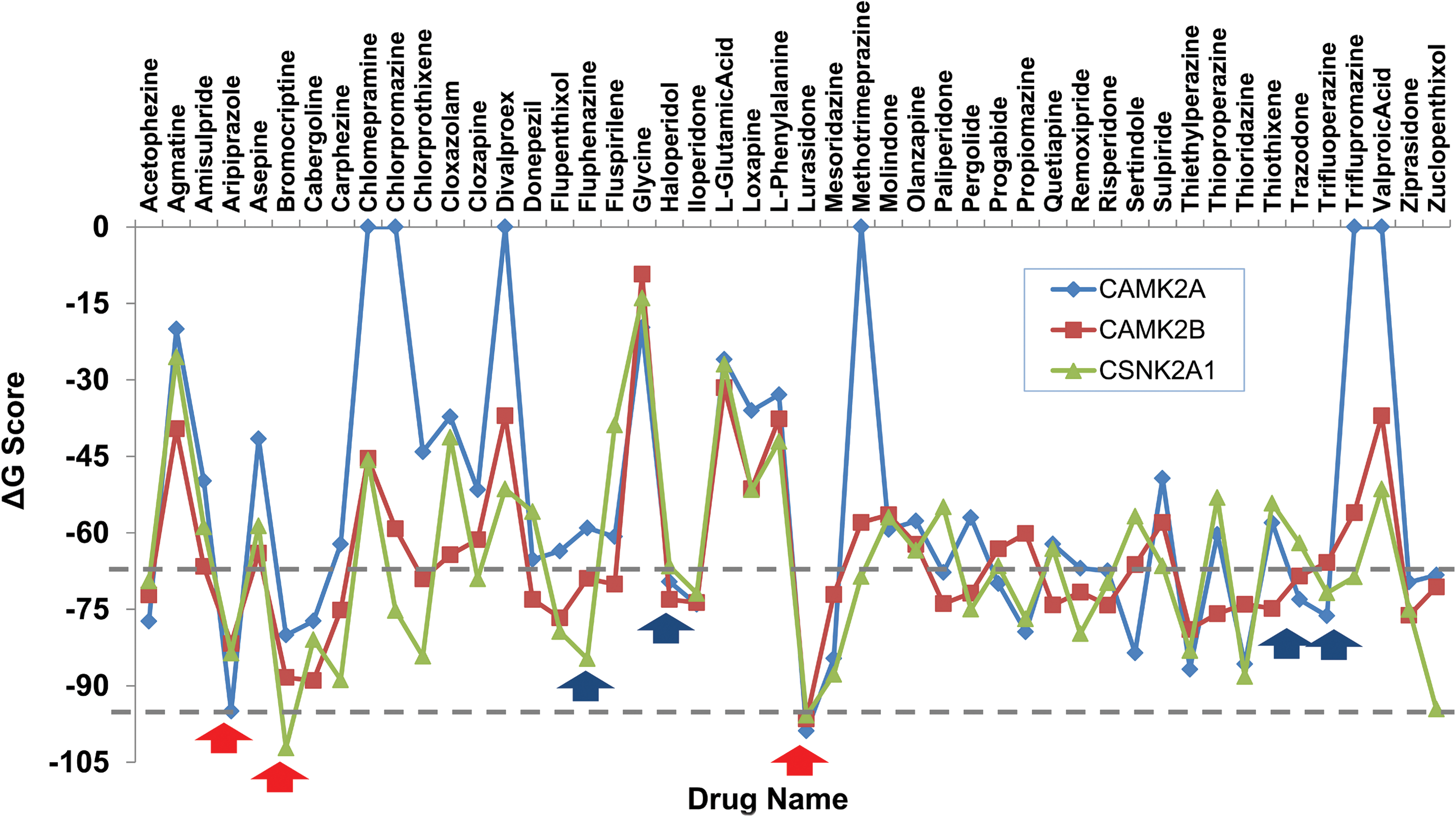
Molecular docking of top three kinases (CSNK2A1, CAMK2A, and CAMK2B) for all available schizophrenia drugs. Prioritization of 48 known schizophrenia-specific drugs based on molecular docking targeting top three proteins kinases as prospective drug targets. Red arrow indicates drugs having higher affinity for all the kinases in the study. Blue arrows depict a respected docking score and reveal the polypharmacological properties of few bridge drugs available in the market.
Discussion
A complex disorder is the result of interplay between disease and non-disease genes. Such interactions of genes often follow unique combinations and patterns based upon their presence in different pathways and tissues (Craig, 2008; Janssens et al., 2008). In this study, we have tried to identify the respective pattern of disease gene interactions in schizophrenia by constructing two different networks: one specific to co-expression information among known schizophrenia disease genes (SDG) named DGNet, and another by incorporating the co-expression information of non-disease gene (NDG) as well along with SDG, which we referred to as (DG+NDG)Net.
In order to resolve the complexity of neuropsychiatric disorders such as schizophrenia, we propose that it is essential to identify the common disease elements in various tissues across the brain. With the available SDG, we identified disease gene co-expression networks for twenty different tissues in human brain. Further, a subset of these networks was identified as common interacting network of SDG. The common network was validated with various computational analyses in order to establish its importance in disease process. We observed that majority of the SDG interactions were present in hypothalamus. Earlier studies have also reported the association of hypothalamus with schizophrenia, though its specific role in disease etiology is still not clear (Hans-Gert et al., 2010; Klomp et al., 2012; Tognin et al., 2012). In view of this, we suggest that the hypothalamus plays a crucial role in understanding the tissue specificity of the disease.
Topologically enriched genes are believed to be important regulatory elements in any biological network (Jeong et al., 2001). Most of the hub genes in the common interacting network were identified with high centrality value in both the tissue specific DGNet (hypothalamus) and the overall (DG+NDG)Net. Our data strongly reflect that important candidate genes for schizophrenia can be prioritized and detected based upon their co-expression and interaction patterns in all the brain tissues. Knowing that human HK genes are important to maintain normal biological functions however, ubiquitous expression of HK genes in both normal and disease tissues downgrade their prominence for further disease etiology (Butte et al., 2001). NHK genes are tissue specific, and alterations in NHK genes have repeatedly been shown to cause disease, thereby potential targets for drugs in any disease (Dezso et al., 2008). Perhaps an overall increase of HK genes in the (DG+NDG)Net indicated the adequacy of DGNet to represent the important NHK genes for schizophrenia. The local arrangement of these NHK genes in common interacting network of SDG is also important for the functionality of many candidate pathways.
We identified amphetamine addiction (KEGG ID: 5031) and dopaminergic synapse (KEGG ID: 4728) as most significant pathways enriched by SDG in the common DGNet. Undoubtedly, drug abuse has been a common concern in schizophrenia, though its role as a cause or as an effect of disease is still not clear (Bühler et al., 2002; Dixon et al., 1991). Prolonged use of amphetamine, cocaine, and similar drugs enhance dopamine levels in brain and lead to symptoms which resemble positively with those in schizophrenia (Jacobs and Silverstone, 1986). Chemical imbalance in brain due to malfunctioning of the dopaminergic system has been shown as a fundamental cause of many neurological diseases. A highly debatable hypothesis—“Dopamine hypothesis”, proposes dopamine to be a causative agent for schizophrenia, and drugs that increase dopamine activity worsen schizophrenic's symptoms (Abi-Dargham, 2004; Howes and Kapur, 2009; Lau et al., 2013). It has also been established that drugs like amphetamine increase dopamine activity, which in turn is the key contributing factor for the etiology of schizophrenia.
However, it is not clear how the corresponding pathways of these molecules crosstalk with each other in the disease process (Boileau et al., 2013; Seeman, 2013; Sunday, 2011). We speculated that there should be a set of interacting SDG that share common pattern of interactions across diverse pathways associated with schizophrenia. We mapped down a subset of interactions for dopaminergic synapse (KEGG ID: 4728) and amphetamine addiction (KEGG ID: 5031) pathways from common interacting network of SDG (Fig. 7). We observed that a group of HK and NHK genes together exhibited a similar pattern of interactions in both pathways. Co-expression pattern between one of the HK gene, calmodulin 1 (CALM1) and two separate families of NHK genes; calmodulin-dependent protein kinase (CAMK2A and CAMK2B) and NMDA receptors (GRIN2A and GRIN2B) were found to be common not only in the above two pathways but also in other pathways such as calcium signaling (KEGG ID: 4020), neurotropin signaling (KEGG ID: 4722), and long-term potentiation (KEGG ID: 4720) (data not shown). Hence, we conclude that candidate pathways are interlinked in schizophrenia through a common co-expression distribution of SDG.
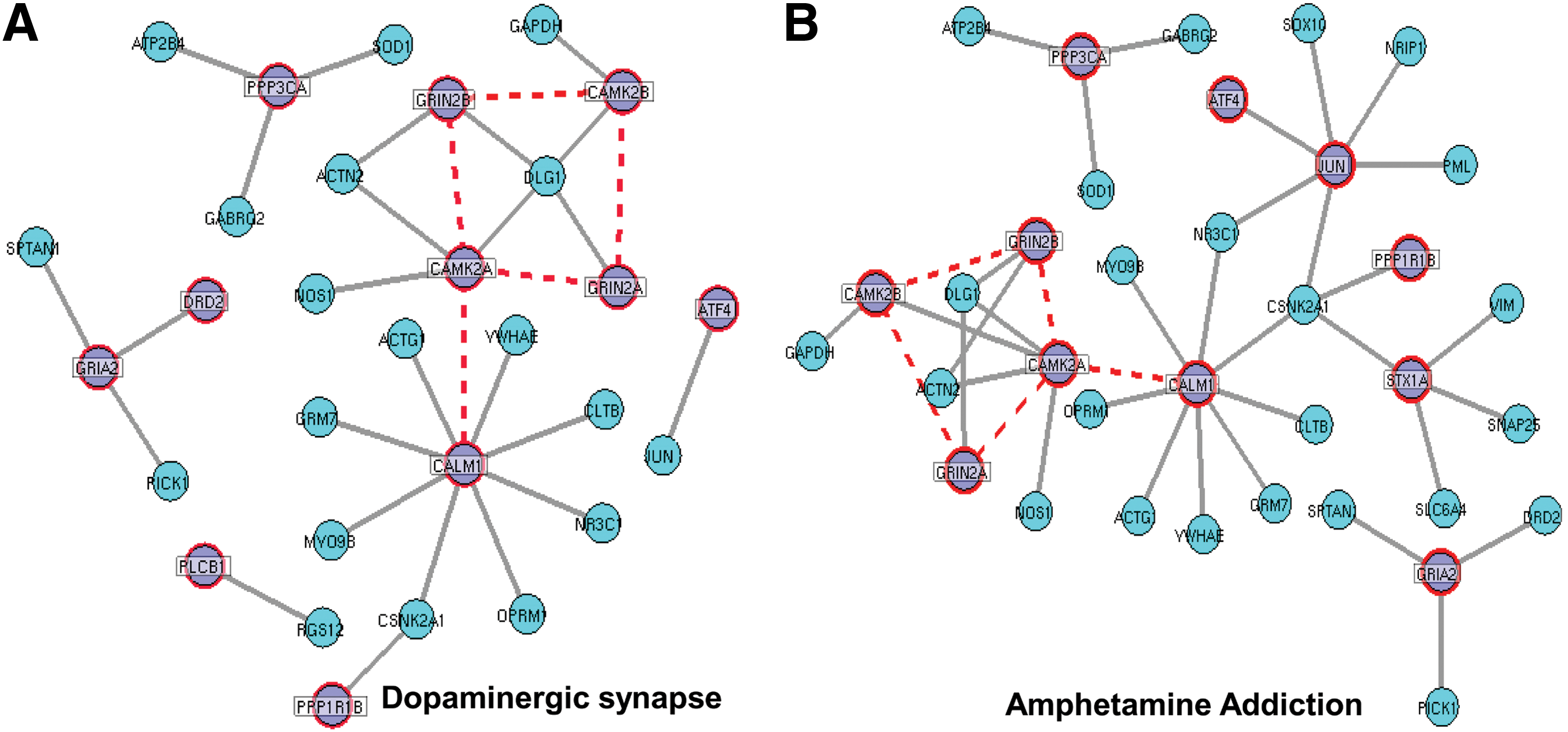
Pathway specific interactions pattern of SDG in common interacting DGNet. A subset of interactions were dragged from DGNet for both
Further enlisting the biological significance of such common co-expression pattern of SDG, we highlighted their positions in KEGG pathway map (Supplementary Fig. S3). It was observed that the genes which exhibited common co-expression pattern are closely interconnected and are mostly involved in the end terminal process of the pathways (highlighted red). However, genes that did not follow any pattern were found to be positioned randomly in the pathway (highlighted yellow and pink). An increase in extracellular concentration of dopamine triggers ‘dopamine-mediated NMDA receptors trafficking’ systems in brain (Aperia and Greengard, 2006; Gao and Wolf, 2008). NMDA receptors are glutamate-gated cation channels that are involved in the modulation of a synaptic response (Dingledine et al., 1999). Activation of NMDA receptors (GRIN2A and GRIN2B) leads to an influx of calcium (Ca2+) into the postsynaptic region where it activates several signaling cascades. Elevated Ca2+ levels allow activation of calmodulin (CALM1) and the kinase activity of calmodulin-dependent kinases (CAMK2A and CAMK2B) which phosphorylate the transcription factor encoded by cAMP responsive element binding (CREB1) gene. As a result, an induced transcription of cAMP-responsive element (CRE) leads to the dysregulation of cyclic adenosine monophosphate (cAMP) that result in development of various symptoms related to schizophrenia and other neurological disorders (Fig. 8).
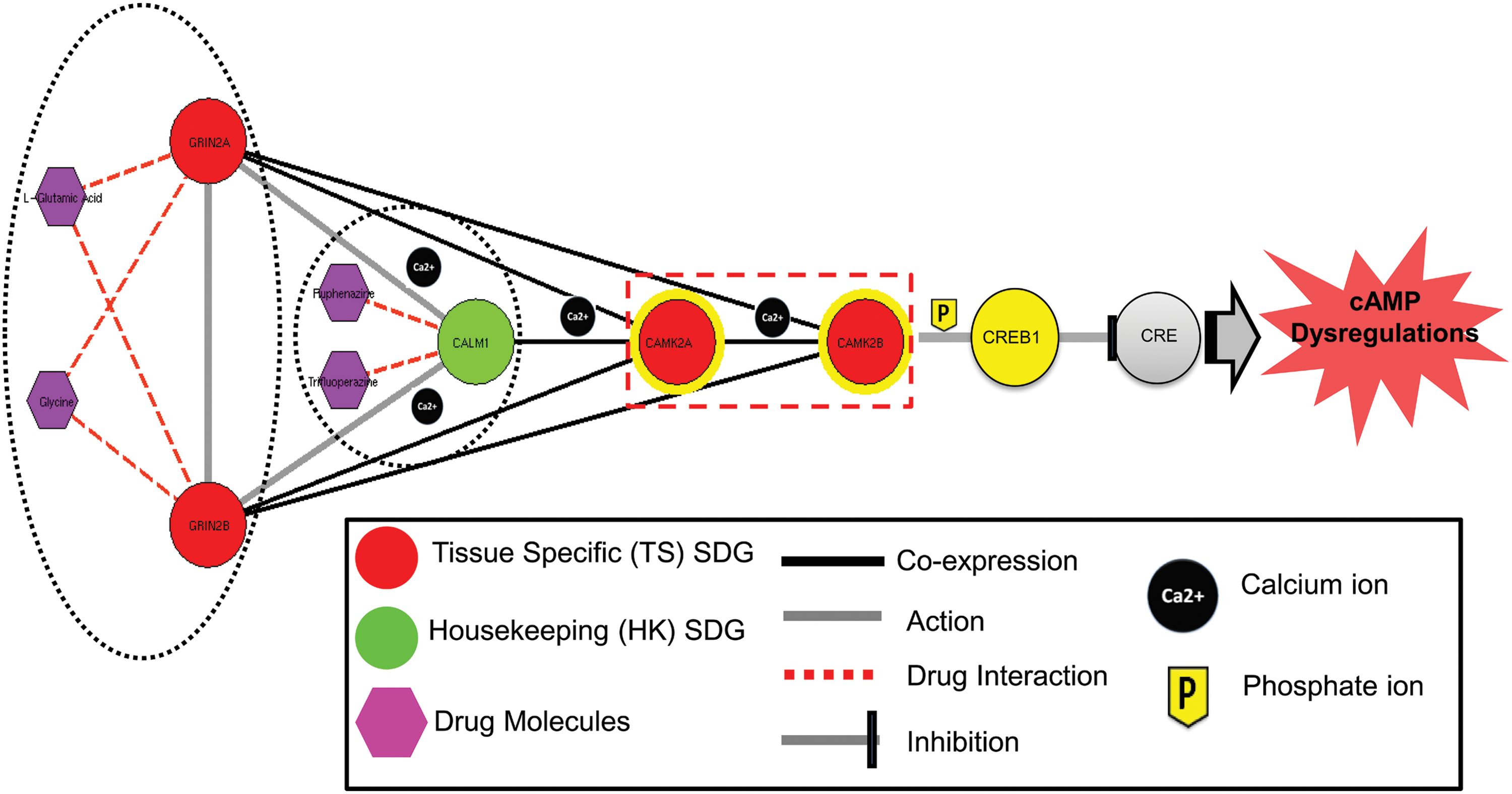
Role of common network elements in cAMP regulation. Common pattern of interactions between SDG play an important role in cyclic AMP regulation process. A schematic representation shows the combined role of major SDG along with other members in downstream cAMP regulation and indicates a central role of calmodulin kinases (CAMK2A and CAMK2B).
Genes that followed a common pattern of interaction are highly organized at functional level and thus are important candidates as drug targets in schizophrenia. Here, NMDA receptors were seen to share common binding site for schizophrenic drugs such as L-glutamic acid (DB00142), glycine (DB00145), and haloperidol (DB00502) (Supplementary Fig. S4). Earlier studies have shown that drug targeted network in schizophrenia is mostly centered with disease genes from the dopamine and serotonin family (Potkin et al., 2010) that also appeared as an important common SDG in majority of the pathways in our study (DRD2 and HTR2A). However, the pharmacological field is still limited by drugs for proteins such as calmodulin-dependent kinases (CAMK2A and CAMK2B) that are key mediators for the secondary messenger-mediated signal transduction in the disease pathways. It is well established that abnormal protein phosphorylation by kinases is an underlying cause or a consequence in several diseases. Hence, apart from G-protein couple receptors, protein kinases have also been considered as important drug targets in the last decade (Badrinarayan and Sastry, 2013; Cohen, 2002; McGuire et al., 2014). We identified three different protein kinases (CSNK2A1, CAMK2A, and CAMK2B) as chief candidate SDG in both the topological and pathway based analysis. Surprisingly, none of these have been targeted by any of the available schizophrenia-specific drugs designed so far.
Therefore, we carried out docking studies of the three protein kinases with known schizophrenic drugs. It was observed that haloperidol features similar binding affinities with all the three kinases apart from targeting GRN2B, DRD2, and HTR2A. We also observed that few bridge drugs such as trazodone and trifluoperazine show similar interaction energetics with all three kinases in addition to the known targets such as SLC6A4, HTR2A and CALM1, DRD2, respectively (Supplementary Fig. S4). On the other hand, the drugs lurasidone, aripiprazole, and bromocriptine also appear to bind significantly with all three kinases. These observations reflect the therapeutic potential of protein kinases for further drug development in schizophrenia.
Association of the available marketed antipsychotic drugs with systemic side effects (Divac et al., 2014; Werner and Coveñas, 2014) can further guide us to design novel drugs by optimizing off-target binding at early stages of drug development. Given the multifactorial nature of schizophrenia, a systematic analysis of disease genes based on genetic variants also will further help in better understanding of disease pathophysiology.
Conclusions
We constructed a schizophrenia disease gene interaction network based upon the co-expression statistics of known disease genes that are common in twenty different zones of human brain tissue. We highlighted the common network elements based on the rich topological properties of SDG and their common pattern of distribution in disease candidate pathways. Interactions amongst the SDG appeared functionally more relevant than their interactions with other non-disease genes for schizophrenia. We isolated a few bridge drugs and evaluated their binding affinity for some important candidates in the network through molecular docking studies. We also suggested that identification of common interaction patterns of genes in the schizophrenia disease–gene interaction network will help to prioritize the essential targets for future drug discovery endeavours in the treatment of schizophrenia.
Footnotes
Acknowledgments
This work is supported by the Department of Biotechnology (DBT), Ministry of Science and Technology, Government of India. We also thank all anonymous reviewers for their critical comments and suggestions that helped us to improve this article.
Author Disclosure Statement
Both authors declare that they have no competing financial interests.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.