
Abstract
Abstract
Leishmania donovani is a kinetoplastid protozoan that causes a severe and fatal disease kala-azar, or visceral leishmaniasis. L. donovani infects human host after the phlebotomine sandfly takes a blood meal and resides within the phagolysosome of infected macrophages. Previous studies on host–parasite interactions have not focused on Leishmania organelles and the role that they play in the survival of this parasite within macrophages. Leishmania possess glycosomes that are unique and specialized subcellular microbody organelles. Glycosomes are known to harbor most peroxisomal enzymes and, in addition, they also possess nine glycolytic enzymes. In the present study, we have carried out proteomic profiling using high resolution mass spectrometry of a sucrose density gradient-enriched glycosomal fraction isolated from L. donovani promastigotes. This study resulted in the identification of 4022 unique peptides, leading to the identification of 1355 unique proteins from a preparation enriched in L. donovani glycosomes. Based on protein annotation, 566 (41.8%) were identified as hypothetical proteins with no known function. A majority of the identified proteins are involved in metabolic processes such as carbohydrate, lipid, and nucleic acid metabolism. Our present proteomic analysis is the most comprehensive study to date to map the proteome of L. donovani glycosomes.
Introduction
T
Peroxisomes, along with glyoxysomes of plants, Woronin bodies of fungi, and glycosomes of kinetoplastid parasites, belong to the quasi-ubiquitous microbody family of subcellular organelles. Microbodies have a protein-rich matrix bounded by a single membrane and are present in all cells having a nucleus. Peroxisomes contain diverse metabolic pathways depending upon the cell type, and the metabolism can be easily amended so that it becomes essential for cell survival under different environmental conditions (O'Connell et al., 2012). In multicellular organisms, peroxisomes play a crucial role in postnatal development and longevity, but at the cellular level, they are not essential for cell viability and functioning (Van Veldhoven and Baes, 2013). In animal cells, peroxisomes contain enzymes for lipid biosynthesis, ß-oxidation of fatty acids, glyoxylate, and amino acid metabolism (Wanders and Waterham, 2006). Plant peroxisomes contain enzymes for the oxidative photosynthetic carbon cycle, oxidation, and biosynthesis of the plant hormones (Palma et al., 2009). In fungi and yeast, peroxisomes have pathways for oxalate synthesis and the metabolism of methanol, amines, and alkanes (Palma et al., 2009; van der Klei and Veenhuis, 2006). The proteome of microbody organelles varies between species, different tissues, and developmental stages. Proteomic analyses of yeast, plant, and animal peroxisomes have been reported indicating that the environmental conditions determine the qualitative and quantitative protein content in the peroxisomes (Hu et al., 2012; Reumann, 2011; Saleem et al., 2006).
Microbodies do not contain endogenous DNA, and all proteins are encoded by nuclear genes (Subramani, 1993). These proteins are synthesized in the cytoplasm and imported post-translationally into peroxisomes (Hasan et al., 2013). In different phyla, process of peroxisome biogenesis, protein targeting signals, and import mechanisms are highly conserved. Two major pathways play an important role in peroxisomal matrix and membrane proteins acquisition. The first pathway, utilized by many proteins, depends on the conserved peroxisomal targeting sequence type 1 (PTS-1), which is located at the extreme carboxyl terminus of a protein. This motif consists of three amino acids related to –SKL and its conserved variants. A few peroxisomal proteins have an amino-terminally located peroxisomal targeting sequence type 2 (PTS-2) motif. This signal is a bipartite signal consisting of nine amino acids; the consensus sequence can be defined as [RK][LVI]X5[HQ][LA] (Petriv et al., 2004). However, a few proteins targeted to peroxisomes may not possess any signal. Although PTS-1 and PTS-2 motif patterns are apparently well conserved in different phyla, correct prediction of proteins with peroxisomal targeting sequences is a challenging task. In silico predictions can be nonetheless done using computational methods; such analyses have been shown to be highly accurate in different systems (Hawkins et al., 2007; Kunze et al., 2011; Lingner et al., 2012; Opperdoes and Szikora, 2006).
A prominent cellular character relating to the evolution of the kinetoplastid parasites is that they possess microbody organelles called glycosomes that are so named because they contain the nine enzymes of the glycolysis pathway (Opperdoes and Borst, 1977). Glycosomes possess protein targeting machinery similar to peroxisomes and contain pathways that include key energy generating pathways such as glycolysis and fatty acid ß-oxidation (Opperdoes and Michels, 1993). In fact, compartmentalization of these pathways into glycosomes helps the parasites survive brief periods of anaerobic conditions during the intracellular stages. The number of glycosomes is considerably fewer in the amastigote stage as compared to the promastigote form (Opperdoes and Coombs, 2007). Recent genetic studies have demonstrated that correct targeting of proteins to the glycosome, especially enzymes of the glycolytic pathway, is crucial for parasite viability (Furuya et al., 2002; Guerra-Giraldez et al., 2002; Moyersoen et al., 2003).
Comparative proteomic analysis of glycosomes in T. brucei brucei by liquid chromatography-electrospray ionization-quadrupole-time of flight-mass spectrometry (LC-ESI-Q-TOF-MS) and by three-dimensional liquid chromatography–tandem mass spectrometry (3D-LC–MS/MS) have been previously reported (Vertommen et al., 2008). The proteomic analysis of glycosomal membranes has identified 464 proteins from Leishmania tarentolae promastigotes (Colasante et al., 2013). Recently the glycosome proteome of the procyclic form of T. brucei was reported wherein glycosomes were enriched using epitope tagging and magnetic bead enrichment. SILAC based quantitative proteomics was found to be helpful in determining the glycosomal proteins with a high-confidence within this proteome profiling report (Guther et al., 2014).
In the current study, we have carried out an analysis to map the proteome of enriched glycosomes from L. donovani promastigotes. This study resulted in the identification of 1355 proteins from an L. donovani glycosome preparation. We have further analyzed these proteins for the presence of either PTS-1 or PTS-2 motifs and have identified 71 predicted PTS-1 and 36 predicted PTS-2 containing proteins carrying putative peroxisome targeting sequence. Our study provides novel insights into the proteomic composition of glycosomes with respect to its function and biogenesis.
Materials and Methods
Cell culture
Leishmania donovani (MHOM/IN/80/Dd8) promastigotes were cultured in Dulbecco's Modified Eagle Medium (DMEM) medium with 10% FBS at 25°C. The cells in logarithmic stage were harvested and washed with phosphate-buffered saline by centrifugation at 1500 rpm for 5 min at 6°C. This procedure was repeated six times to get rid of any contaminating serum proteins used in cultivation media. The promastigote cells were counted using a Neubauer chamber and 2×109 cells were used for glycosome preparation.
Glycosome preparation
The L. donovani promastigote cell pellet was resuspended in a hypotonic solution [2 mM EGTA, 2 mM DTT, and protease inhibitor cocktail, pH 7.4]. Swollen cells were lysed by passing the suspension ten times through a 27-gauge needle. The cell lysate was made isotonic by addition of 4× assay buffer [50 mM HEPES pH 7.4, 0.25 M sucrose, 1 mM ATP, 1 mM EGTA, 2 mM DTT, and protease inhibitor cocktail], and centrifuged at 3000 g for 10 min to remove intact cells and nuclei. The post nuclear supernatant was fractionated on a linear sucrose gradient [20%–70% sucrose in 25 mM HEPES, pH 7.4], at 218,000 g for 6 h at 4°C in a Beckman L-80 centrifuge using a SW41Ti rotor (Ilgoutz et al., 1999). Western blot analysis of the density gradient fractions (0.4 mL) carried out with anti-L. major hexokinase antibodies revealed that this marker enzyme was present in the 50%–60% fraction of sucrose. This fraction corresponded to the buoyant density of glycosomes (1.2–1.25 g/cm3) and was comparable with the buoyant density of glycosomes reported earlier (Hart and Opperdoes, 1984). Furthermore, the hexokinase activity was confined preponderantly to these fractions, as demonstrated by the enzyme assays. The protein concentration of each fraction was assessed by the Bradford method and was plotted as a function along with the enzyme activity (Supplementary Fig. S1; supplementary material is available online at www.liebertonline.com/omi). The proteins in each fraction were precipitated with an equal volume of a chloroform–methanol mixture (1:1). The workflow utilized for the isolation and purification of L. donovani glycosomes is outlined in Figure 1.

Workflow for glycosome purification and enrichment from L. donovani promastigotes. Promastigotes of L. donovani were cultured and used for the isolation of glycosomes. The cells were resuspended in hypotonic buffer, followed by lysis of the promastigotes by passing the suspension through a narrow gauge syringe. This was followed by brief centrifugation at 3000 g, followed by sucrose density gradient centrifugation at 39,000 rpm for 6 h in an ultracentrifuge. The glycosomes sedimented at the interphase of 2 M and 1.75 M sucrose on the sucrose density gradient.
Protein isolation and fractionation
The purified glycosome fractions were resuspended in 0.5% SDS and sonicated for six cycles at 50% output, as described previously. The glycosomal lysate was centrifuged at 12,000 rpm for 10 min and the supernatant was used for further proteomic analysis. Three hundred micrograms of glycosomal proteins were separated on a 10% SDS-PAGE gel. Individual protein bands were cut from the gel and each band was subjected to reduction (5 mM dithiothreitol) and alkylation (20 mM iodocetamide), followed by in-gel trypsin digestion (1:20 trypsin; weight to weight ratio of trypsin to protein) for 12 h at 37°C. The peptide extraction was carried out, and peptide fractions were vacuum dried. Additionally, the glycosomal lysate was subjected to in-solution trypsin digestion (1:15 trypsin) for 12 h at 37°C. This was followed by fractionation of tryptic peptides using strong cationic exchange (SCX) chromatography as described previously (Pawar et al., 2012; 2014). The peptide fractions were completely dried and reconstituted in 40 μL of 0.2% formic acid.
LC-MS/MS analysis
LC-MS/MS analysis was carried out for a total of 23 fractions obtained from two tryptic digestion protocols (i.e., the ‘in-gel’ and ‘in-solution’ methods), as described above. For further fractionation, a desalting column (5 μ 100 Å Magic C18, Michrom Bioresources) and an analytical column (5 μ 100 Å Magic C18, Michrom Bioresources) were connected to Agilent's 1200 Series Nanoflow LC constituting a reverse phase liquid chromatography (RP-LC) system. The RP-LC was connected on-line to the LTQ-Orbitrap Velos ETD mass spectrometer (Thermo Electron, Bremen, Germany). The nanospray source was fitted with an emitter tip 8 μm (New Objective, Woburn, MA) and a voltage of 2 kV was applied. Peptide samples reconstituted in HPLC solvent A (0.1% formic acid) were loaded on the trap column and washed for 5 min with 97% HPLC solvent A and 3% solvent B (90% ACN in 0.1% formic acid). The peptide separation was carried out using a linear gradient of 7%– 30% solvent B for 53 min at a constant flow rate of 0.4 μL/min. Data was acquired using the Xcalibur 1.2 (Thermo Electron, Bremen, Germany). In the scan range of m/z 350 to 1800 Da, the twenty most abundant ions were selected for fragmentation. The acquired ions were excluded for 30 sec. Target ion quantity for FT full MS was 5×105 and for MSn was 2×105. MS was acquired with the FT analyzer at the resolving power of 60,000. MS/MS was carried out in HCD mode with a resolving power of 15,000 at 400 m/z. The lock mass option was enabled for accurate mass measurements. Polydimethylcyclosiloxane ions were used for internal calibration.
Proteomic data analysis
A non-redundant (nr) protein database of the BPK282A1 strain of L. donovani (n=8032) [database available from NCBI (http://www.ncbi.nlm.nih.gov) as of August 10, 2013] was used for database dependent searches. The Sequest search engine was used to search L. donovani glycosome mass spectrometry data and searches were submitted via the Proteome Discoverer software (version 1.3). The search parameters used for database dependent searches were as follows: a) trypsin as a proteolytic enzyme (with up to one missed cleavage); b) peptide mass error tolerance of 20 ppm; c) fragment mass error tolerance of 0.1 Da; d) oxidation of methionine as variable modification and carbamidomethylation of cysteine as fixed modification. Sequest peptide data were extracted with a 1% false discovery rate (FDR) threshold and unique peptide data obtained after protein database dependent search were used for further analysis. Quantitation of proteins was carried out based on intensity-based absolute quantification (iBAQ) and normalized spectral abundance factor (NSAF)-based spectral counting. For calculation of iBAQ, the MS1 level intensities of spectral matches of all peptides identified for a protein are summed and normalized to the number of theoretical tryptic peptides (Schmidt et al., 2011), whereas NSAF quantifications are derived by taking the ratio of number of spectral matches for an identified protein to the number of theoretical tryptic peptides (Zybailov et al., 2006).
Bioinformatics analysis
The proteins identified in our current analysis were categorized into groups based on the biological process in which they are involved (e.g., cell cycle proteins). These analyses were performed in compliance with Gene Ontology [GO] standards. The TriTrypDB database (Aslett et al., 2010) was used for defining the transmembrane domains and signal peptides. The TriTrypDB database uses the TMHMM (Krogh et al., 2001) and the SignalP (Petersen et al., 2011) software for predicting transmembrane domains and signal peptides, respectively. Additionally, analysis was carried out using the motif search program available at GeneDB (Logan-Klumpler et al., 2012) to identify peroxisomal targeting sequences type-1 and type-2 in the putative glycosomal proteins identified in this study. For the search, the [ASCGPNYTV] [KNRHQDS][LMVAIF] motif was used to identify proteins having the PTS-1 sequence, while the [RKHQ][VLIWFY]X5[HKQR][ILVYAF] sequence was used to identify proteins with the PTS-2 motif. These PTS-1 and PTS-2 sequences identified in our analysis were manually checked and verified. PTS predictions analysis was performed using the Peroxisome database [www.peroxisomedb.org].
Proteomic data availability
To make our observations publicly available and accessible, the complete set of mass spectrometry data generated from this study have been deposited to the ProteomeXchange Consortium (http://www.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD000724 (Vizcaino et al., 2013).
Results
Summary of proteomic data
The aim of this study was to carry out a detailed proteomic analysis of enriched glycosomal fraction from L. donovani promastigotes using high resolution mass spectrometry. The strategy used for proteomic profiling of L. donovani glycosomal fraction is outlined in Figure 2. The 23 LC-MS/MS data files that were acquired in the present study were used for the current analysis. This dataset included 142,621 MS/MS spectra that were searched using the Sequest search algorithm against the recently available L. donovani protein database. In all, 73,236 peptide spectrum matches (PSMs) that led to the identification of 4022 unique peptides at 1% FDR cutoff were identified from protein database searches. This in turn resulted in the identification of 1355 unique proteins in the glycosomal fraction of L. donovani. Supplementary Tables S1 and S2 provide a complete list of proteins and peptide identified in protein database searches. Of these, 566 proteins were ‘hypothetical’ proteins, six proteins were ‘unnamed products’ and 435 proteins were grouped as ‘putative glycosomal’ proteins. This ‘putative glycosomal’ grouping was based on presence of PTS motif, reports of these proteins as true glycosomal or peroxisomal proteins from literature, and the UniProt database.

Workflow for sample processing, fractionation, and proteomic analysis of L. donovani glycosomes. L. donovani promastigotes were used for the isolation and purification of glycosomes for proteomic analysis as indicated. Proteins from the glycosome were extracted using SDS and subjected to SDS-PAGE or digested with trypsin and subjected to strong cation exchange (SCX) chromatography. LC-MS/MS analysis of the digested gel bands or SCX fractions was carried out on a high resolution mass spectrometer. The mass spectrometry data were analyzed against a protein database of L. donovani.
The proteomic profiling reports for plant, animal, and kinetoplastid data were also considered while grouping these 435 proteins as putative glycosomal proteins. For example, various heat shock proteins, RNA helicase, GTP binding protein, and cyclophilin were reported to be putative glycosomal proteins on the basis of the peroxisomal profile of Arabidopsis thaliana peroxisomes (Reumann et al., 2009) or “Sec proteins” involved in protein transport and secretion were found to be peroxisomal by co-localization studies (Perry et al., 2009). Quantitation of proteins was carried out using MS1 level precursor intensity (iBAQ) and spectral count (NSAF) based relative expression of proteins identified in this study. The quantitative data is also provided in Supplementary Tables S1 and S2. The data indicate that there is a good correlation between the iBAQ values and proteins that are of glycosomal origin. The enzymes or proteins that are reported to be glycosomal by earlier studies, had very high iBAQ values, such as glyceraldehyde 3-phosphate dehydrogenase, hexokinase, and ATP-dependent phosphofructokinase.
Peroxisomal Targeting Sequence type-1 (PTS-1) and Peroxisomal Targeting sequence type-2 (PTS-2) containing proteins in purified glycosomes
Based on earlier targeting studies (Blattner et al., 1992; Sommer et al., 1992) and analysis performed in silico (Opperdoes and Szikora, 2006), the trypanosomatid PTS-1 and PTS-2 motifs appear to have very divergent sequences than those of plants and humans. Based on glycosomal proteins reported by in silico analysis (Opperdoes and Szikora, 2006), we have used [ASCGPNYTV][KNRHQDS][LMVAIF] motif to search proteins with putative PTS-1 signal in proteins identified from enriched glycosome fraction of L. donovani promastigotes. Using this, we found 71 proteins having the predicted PTS-1 motif in our present proteomic analysis. Supplementary Table S3 lists glycosomal proteins containing putative PTS-1 sequences identified in the current proteomic analysis of L. donovani glycosomes. Some of the putative proteins with unusual PTS-1 motifs, which are unlikely to represent true PTS, were included in the list as they are known to participate in pathways that are operative in glycosomes or peroxisomes. These include proteins such as ubiquinone biosynthesis methyl-transferase [having -NSA as a putative motif] or peroxisome targeting signal-1 receptor [-GHV as a putative motif]. Some of the proteins included in the list have a very high alkaline pI (in the range 8.8–10.2), which is a hallmark of glycosomal proteins (Misset et al., 1986). There is a probable risk of labeling such unusual non-canonical motifs as PTS-1 signals but such in silico analysis can be further confirmed only by in vivo targeting studies in the parasite.
Similarly, there is very little variation in the nine-mer PTS-2 sequences reported from mammalian, plants, and yeast, especially at positions 1, 2, 8, and 9, and the consensus motif is described as [RK][LVI]X5[HQ][LA] (Petriv et al., 2004). We have selected the [RKHQ][VLIWFY]X5[HKQR] [ILVYAF] motif, based on a list of various PTS-2 sequences detected in kinetoplastids (Opperdoes and Szikora, 2006), for identification of proteins with a PTS-2 sequence in our present proteome analysis of L. donovani glycosomes. Supplementary Table S3 shows a list of putative glycosomal proteins containing putative PTS-2 sequences identified in the current proteomic analysis of L. donovani glycosomes. In our glycosomal proteome list, we found 36 proteins having this predicted PTS-2 motif. These 107 proteins with predicted and putative PTS motifs were analyzed for subcellular targeting potential using the PreoxisomeDB database. The ‘Target Signal Predictor’ reported that all the 107 proteins had at least one single hit matching with the database, indicating positive peroxisomal targeting potential of the Leishmania PTS sequences.
Metabolic enzymes encompassed in glycosomes
Glycosomes are metabolic hubs within the Leishmania parasite and contain a pool of key metabolic enzymes required for energy metabolism. Glycosomes contains enzymes involved in various metabolic pathways such as glycolysis, pentose phosphate pathway, β-oxidation of lipids, sterol biosynthesis, and nucleic acid metabolism. The enzymes identified in our proteomic analysis of L. donovani glycosomes are described below, along with their role in the metabolic pathways (Supplementary Table S4).
Glycolysis
Glycolysis is the central pathway for energy metabolism that utilizes carbohydrates as a source of energy in most living organisms. We identified nine key enzymes involved in glycolysis in the glycosomal fraction. We identified an important glycolytic enzyme, ATP-dependent 6-phosphofructokinase (LDBPK_292620). It is a 486 amino acid long protein (Fig. 3A) that mediates the phosphorylation of fructose-6-phosphate to fructose-1,6-bisphosphate and contains a putative C-terminal PTS-1 sequence “-SKV”. We have identified 21 unique peptides (Fig. 3B) of ATP-dependent 6-phosphofructokinase in our proteomic analysis. Figure 3C shows the representative MS/MS spectrum of one peptide (TFGFQTAVEQAVNAVR) for the PTS-1 containing ATP-dependent 6-phosphofructokinase identified in our analysis.
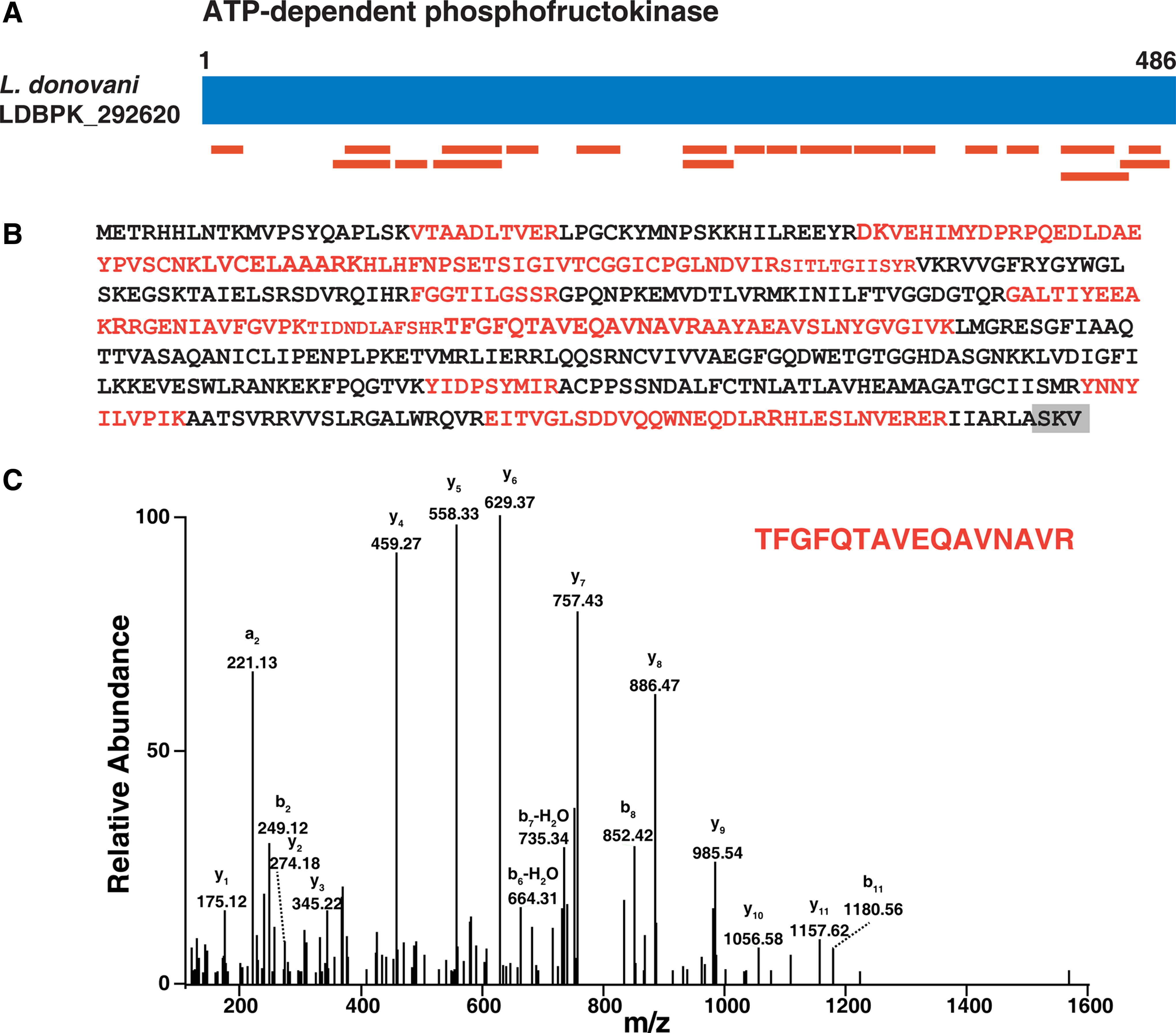
Identification of PTS-1 containing protein ATP-dependent phosphofructokinase.
We also identified L. donovani hexokinase (LDBPK_210300) a 486 amino acid-long protein (Fig. 4A), which is a rate-limiting enzyme in the glycolytic pathway and is involved in the phosphorylation of glucose to glucose-6-phosphate. Hexokinase contains an N-terminal PTS-2 sequence “-RVNNLLSHI-” which is conserved in L. major, L. mexicana, and L. infantum. As described by Petriv et al. (2004), the amino acid at the ninth position in the PTS-2 motif is highly conserved and is either leucine or valine. But the motif from leishmanial hexokinase has isoleucine at the ninth position, which is not reported in any PTS-2 sequences described to date. We have identified 22 unique peptides (Fig. 4B) for hexokinase in our proteomic analysis. Figure 4C shows the representative MS/MS spectrum of one peptide (ATGVYYALDLGGTNFR) for PTS-2 containing hexokinase identified in our analysis.
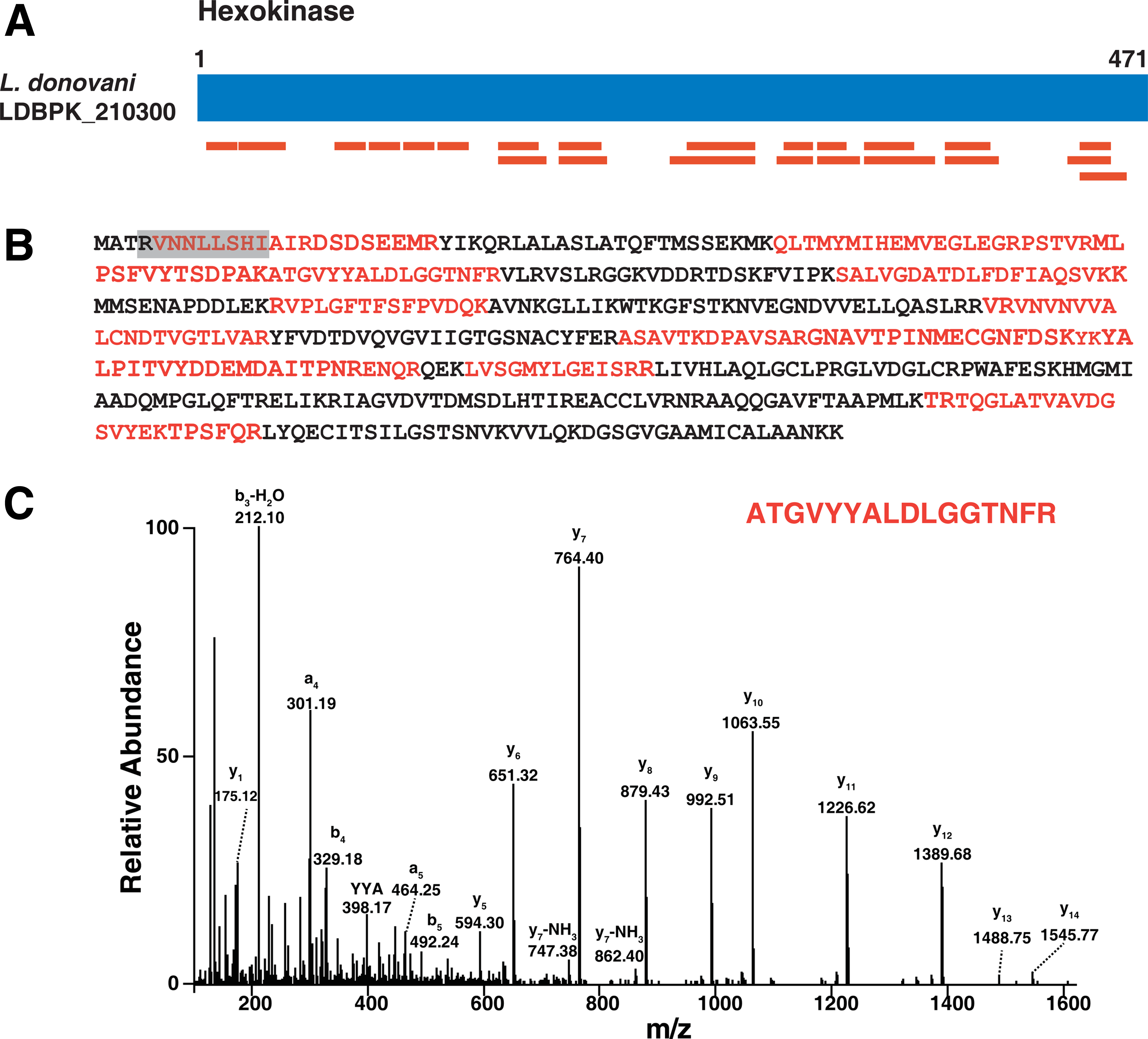
Identification of PTS-2 containing protein hexokinase.
Pentose phosphate pathway
The pentose phosphate pathway, also known as the hexose monophosphate shunt, is a pathway that generates reducing agents such as NADPH and pentose sugars essential for nucleic acid and fatty acid biosynthesis. We identified the six enzymes involved in this pathway. We identified 6-phosphogluconate dehydrogenase (LDBPK_353390), which catalyzes the conversion of 6-phosphogluconate to ribulose-5-phosphate, and have identified two unique peptides for this protein in our proteomic analysis. We also identified transketolase (LDBPK_242150) which catalyzes the conversion of erythrose-4-phosphate and xylulose-5-phosphate to glyceraldehydes-3-phosphate and fructose-6-phosphate and contains a C-terminal PTS-1 sequence “SKM”. We have identified four unique peptides for transketolase in our proteomic analysis.
Lipid metabolism
The β-oxidation of fatty acids generates energy in the form of ATP, and in most organisms this pathway takes place in the peroxisomes, including even in the leishmanial glycosome (Hart and Opperdoes, 1984). The glycosomal β-oxidation pathway of fatty acids is a major source of energy for the parasite in the amastigote stage (Berman et al., 1987). Although previous proteomic analyses failed to detect any enzymes in this pathway in T. brucei (Vertommen D et al., 2008), in our proteomic analysis of the L. donovani glycosomal fraction, we have identified six unique peptides for the enoyl-CoA isomerase enzyme (LDBPK_332740) that catalyzes the hydration of acyl-CoA to 3-hydroxyacyl-CoA. We also identified another enzyme involved in β-oxidation, 3-ketoacyl-CoA thiolase (LDBPK_230860), which is involved in carbon bond formation and catalyzes the conversion of acyl-CoA and acetyl-CoA to Coenzyme A and 3-oxoacyl-CoA. 3-ketoacyl-CoA thiolase contains an N-terminal PTS-2 sequence “RVFIVGGHI”. We have identified six unique peptides for 3-ketoacyl-CoA thiolase in our proteomic analysis. It is known that the substrates of β-oxidation are transported into peroxisomes by the peroxisomal ATP binding cassette transporter protein CTS/PXA1/PED3. A homologue of this was found in the glycosomes of L. donovani (LDBPK_310560) has a domain similar to the ATP-binding cassette protein subfamily D, member 2. The peroxisome is also the site for ether–lipid biosynthesis, and we have detected the presence of alkyldihydroxy-acetonephosphate synthase (LDBPK_300120) and dihydroxyacetonephosphate acyltransferase (LDBPK_341170) enzymes in glycosomes. Although it is known that most sterol biosynthesis occurs in peroxisomes and glycosomes, we have not found any peptide that belongs to enzymes involved in cholesterol biosynthesis. The presence of cholesterol in fetal bovine serum that is added to medium for cultivation of the promastigote stage may be the source of cholesterol for the parasite.
Purine and pyrimidine metabolism
Purines and pyrimidines are an important pool of biomolecules as these molecules acts as co-factors, precursors of nucleic acids, secondary messengers, and the energy currency of cells. A de novo purine biosynthesis pathway is absent in Leishmania and the parasite depends on its host for its purine requirements and also possess enzymes for the purine salvage pathway (Boitz et al., 2012). In comparison to purine salvage pathway, Leishmania parasites are prototrophs for pyrimdine and also express the pyrimidine salvage enzymes. In Leishmania and other related parasites, enzymes in purine and pyrimidine metabolism are compartmentalized inside glycosomes (Carter et al., 2008) and most of these enzymes have either the PTS-1 or PTS-2 motif (Boitz et al., 2012).
In our current proteomic analysis of the L. donovani glycosome enriched fraction, we have identified five enzymes of the purine salvage pathway (Supplementary Table S4). One of the proteins detected was xanthine phosphoribosyl transferase (XPRT; LDBPK_210990) which catalyzes the formation of xanthine and 5-phospho-α-D-ribose-1-diphosphate from xanthosine monophosphate and diphosphate. Xanthine phosphoribosyl transferase has an –AKL motif at the C-terminal, which is a potential PTS-1 signal, and this corroborates with its glycosomal localization, as seen in an immuno-localization study (Zarella-Boitz et al., 2004). The other enzyme found in the glycosomal fraction was inosine monophosphate dehydrogenase (IMPDH; LDBPK_191590) which maintains the intracellular guanylate pool (Dobie et al., 2007) and is an important enzyme, as parasites lacking this enzyme show limited growth in culture, but was found to be non-essential for infection in mice (Fulwiler et al., 2011). We have identified 12 unique peptides of inosine monophosphate dehydrogenase (IMPDH) (LDBPK_191590) in our current proteomic analysis of the L. donovani glycosome-enriched fraction. We have identified the PTS for inosine monophosphate dehydrogenase to be the C-terminal PTS-1 tripeptide –AKM. The only enzyme in pyrimidine synthesis that was detected in this proteomic analysis was orotate phosphoribosyltransferase (LDBPK_160560) for which we have identified 11 unique peptides. This enzyme also has a canonical C-terminal PTS-1 tripeptide motif –SKL.
Peroxisome biogenesis proteins
Important receptors that enable transfer of proteins to the peroxisomal matrix using PTS-1 and PTS-2 and various peroxisome assembly proteins were detected in the glycosomal fraction of L. donovani promastigotes (Table 1). ATP binding cassette transporter proteins are associated with various intracellular compartments including peroxisomes and are implicated in the biogenesis of peroxisomal membranes. T. brucei ABC transporters (Yernaux et al., 2006) GAT-1 and -3 orthologues, but not GAT-2, could be found in the glycosomes of L. donovani (LDBPK_270480 and LDBPK_310560). Membrane-bound peroxin-3 is involved in targeting ATP binding cassette transporter to the peroxisomal membrane. A peroxin-3 homologue (LDBPK_180860) was also found along with two ATP binding cassette transporters in the glycosomes.
A majority of the membrane proteins and lipids that are required for the de novo biogenesis of peroxisomes, originate mostly from the endoplasmic reticulum (Agrawal and Subramani, 2013). Vesicles loaded with peroxisomal membrane proteins bud from the ER and fuse with other ER-derived peroxisomal membrane protein vesicles to form a new peroxisome compartment. In mammalian and yeast systems, vesicular transport from the endoplasmic reticulum to peroxisomes involves ADP ribosylation factor-1 (ARF-1) and coat protein complex (COPI) coat-binding proteins. These proteins have also been implicated in vesicular cycling (Lay et al., 2006). Both the coat protein complex (LDBPK_322150) and ARF (LDBPK_312350 and LDBPK_312890) proteins have been detected in the present leishmanial glycosomal preparation. A complete set of proteins essential for heterotypic fusion reactions requires N-ethylmaleimide-sensitive factor (NSF; LDBPK_200820), and soluble NSF attachment protein receptors (SNAREs; LDBPK_352760 and LDBPK_070570) (Lam et al., 2010) were also found in glycosomal fraction. Several Rab proteins, ras-like small GTPase, and GTP binding proteins that are essential regulators of vesicle trafficking (Gronemeyer et al., 2013) were also found in the proteomic analysis of glycosomes. Chaperone T-complex (LDBPK_321060) and small Rab-GTPases proteins known to interact with peroxin-7, were found in the glycosomal fraction.
In yeasts, plants, and animals, peroxin-11 and its variants are known to play an important role in peroxisome division and proliferation (Opalinski et al., 2011). In the L. donovani glycosomal fraction, we could detect the presence of protein Gim5A [putative] encoded by LDBPK_353750; this protein has a peroxin-11 domain. Another important class of proteins in peroxisomal proliferation is large GTPases called dynamins. Dynamins bring about fusion and fission of organelles by forming ring-like structures around the organelle membranes and finally cutting and splitting the membranes (Opalinski et al., 2011). The L. donovani gene LDBPK_292310 that encodes for a GTP binding protein with dynamin domains and this protein was found to be present in glycosomes. However, the interacting partners of dynamin essential for peroxisomal fission, namely fission 1 protein [FIS1] and mitochondrial fission factor [MFF], were not detected in this proteomic analysis.
Mitochondria-derived vesicles also transport cargo from the mitochondria to the peroxisomes, and these vesicles are known to carry ubiquitin-like modifier (small ubiquitin-like modifier protein; SUMO) E3-ligase. The mitochondria-anchored protein ligase (MAPL) carrying vesicles are known to get targeted to peroxisomes (Li et al., 2008). We have found a homologue of this in glycosomes that is encoded by gene LDBPK_181160. The putative protein encoded by this gene shows the presence of a RING domain and E3 ubiquitin-protein ligase. It has been shown that UPS regulates chloroplast biogenesis, particularly by RING-type ubiquitin E3 ligase located in the chloroplast outer membrane (Ling et al., 2012). This protein is reported to be present in the peroxisomal membrane; we could detect a ubiquitin E3 ligase-like protein in the glycosomal fraction. The RING finger peroxins—peroxin-2p, peroxin-10p and peroxin-12p—which aid in matrix protein import also possess ubiquitin-protein ligase activity. This activity is also associated with ubiquitin-dependent regulation of peroxin-5p (El Magraoui et al., 2012). Alignment of these three putative peroxins from L. donovani showed highly conserved zinc binding cysteine residues.
Structural proteins associated with glycosomes
The uniform distribution of peroxisomes in the cytoplasm is attributed to their binding to microtubules (Hammer and Sellers, 2012). Multiple intracellular interactions and functional exchanges between peroxisomes and other organelles are well known phenomena and explain the different variations in peroxisomal metabolism. Interactions between peroxisomes and lipid droplets play an important role in lipid metabolism (Schrader, 2001). These interactions are brought about by microtubules and their associated proteins. Several microtubular proteins, such as α-, β- and γ-tubulin, microtubule-associated protein, and tubulin tyrosine ligase, were detected in our glycosomal preparations. Peroxisomal bidirectional transport is mediated by the microtubule motor proteins kinesin-1; this protein is known to co-localize with peroxisomes (Dietrich et al., 2013). Gene products of LDBPK_190250 and LDBPK_300350 were present in the glycosomal fractions of L. donovani and these proteins have the kinesin motor domain-2, ATPase, and VHS domains that are essential for organelle transport. Besides kinesin and microtubules, other cytoskeletal proteins such as actin and dynein were also detected in the glycosomal enriched fraction of L. donovani.
Non-glycosomal proteins in glycosomal fraction
Subcellular fractionation and purification of an organelle from total cell extracts are known to reduce the complexity in the preparation by essentially removing contaminating organelles and cytoplasmic proteins. This also helps to enrich the low abundant organelle specific proteins. These two aspects make such preparations most suitable for proteomic profiling of cellular organelles. But in this study, we have detected several non-glycosomal proteins co-sedimenting in the glycosomal fractions. These non-glycosomal proteins were mainly histones, ribosomal proteins, chaperones, mitochondrial proteins, and structural proteins, as well as components of the protein synthesizing machinery and ubiquitin-proteasome system (Supplementary Table S5).
Similar non-glycosomal proteins have also been reported in glycosomal proteomic profiling done earlier in Leishmania, and Trypanosoma and also in peroxisomal preparations from plants (Reumann et al., 2009). Ribosomes, initiation and elongation factors, and chaperones co-sediment with peroxisomes or glycosomes, an observation that can be justified by the fact that preparations of intracellular membranes often have attached ribosomal particles (Fujiki et al., 1982). Mitochondrial proteins were also observed in our preparation, as well as in earlier reports. The percent purity of peroxisomes isolated by isopycnic gradient is approximately 95%, and it carries some non-glycosomal proteins of mitochondria and endoplasmic reticulum origin (Alexson et al., 1985; Opperdoes et al., 1984).
Comparison of proteomic profiles of three kinetoplastid parasites
The glycosomal proteome of the procyclic form of T. brucei was reported recently by Guther et al., (2014), in which glycosomes were enriched using epitope tagging and magnetic bead enrichment. This study reported the presence of 3182 proteins in the affinity purified glycosomal fraction. Although this study utilized a preparation with affinity purified glycosomes, it carried several non-glycosomal proteins such as histones, ribosomal proteins, proteins involved in protein biosynthesis, and mitochondrial proteins. SILAC based quantitative proteomics helped Guther et al., (2014) to determine glycosomal proteins with a high-confidence within the glycosome proteome profile of T. brucei. Putative proteins of glycosomes from L. donovani promastigotes identified in this study were compared with the glycosomal proteins from L. tarentolae promastigotes (Colasante et al., 2013) and the procyclic form of T. brucei (Guther et al., 2014). The comparison showed that, out of 1355 proteins of L. donovani, there were 936 proteins common with T. brucei and 265 proteins common with the glycosomal preparation of L. tarentolae. The comparison indicated that profiling of L. donovani glycosomes has identified many unique proteins that have not been detected in proteomic studies of L. tarentolae and T. brucei glycosomes (Supplementary Table S6 and Fig. 5). Some of the important proteins found only in L. donovani glycosomes were glycosomal membrane protein, orotidine-5-phosphate decarboxylase/ orotate phosphor-ribosyltransferase (de novo pyrimidine nucleobase biosynthesis), transketolase (pentose phosphate pathway), glucokinase and ribulokinase (sugar metabolism), 4-coumarate-CoA ligase (biquinone biosynthesis), arginase (intracellular survival), peroxisomal enoyl-coa hydratase and glycerol-3-phosphate acyl transferase (lipid metabolism).
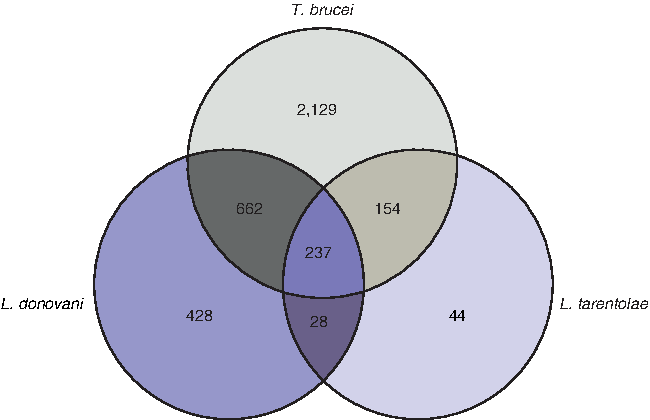
Comparison of glycosomal proteomic data from L. donovani, L. tarentolae, and T. brucei. The comparison of proteins detected in proteomic study of three kintoplastid glycosomal preparation shown by Venn diagram indicates number of orthologues found in all three preparations.
Gene ontology and bioinformatics analysis
Gene ontology (GO) analysis was carried out to classify proteins based on biological processes. The proteins identified in the current proteomic analysis of L. donovani glycosomes were classified based on the biological process in which they are involved (e.g., metabolism). The GO analysis resulted in 615 proteins (45%) being grouped into one of the biological processes (Fig. 6A) and 759 proteins (56%) being grouped based on molecular function (Fig. 6B). In addition, the bioinformatics analysis of L. donovani glycosomal proteins was carried out to identify transmembrane and secreted proteins. Of the 1355 proteins identified in L. donovani, 156 proteins contained transmembrane (TM) domains and 123 proteins contained only signal peptides (SP). Additionally, 117 proteins contained both a transmembrane domain and a signal peptide (Fig. 6C).
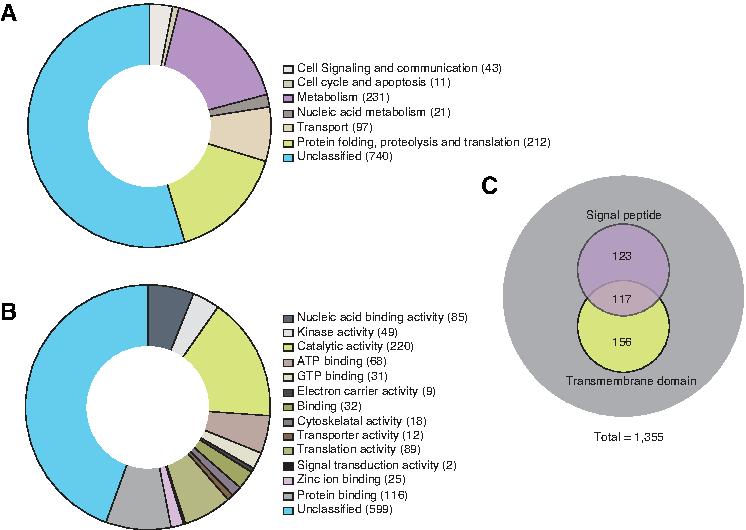
Gene ontology based bioinformatics analysis of proteins identified in L. donovani glycosome.
In addition, L. donovani has many genes that are functionally uncharacterized and do not have sequence similarity with other known genes in protozoa. Many of these proteins could be potentially interesting targets for drug and vaccine development. We have identified various hypothetical proteins in the glycosomal fraction that have no known function assigned. A list of 566 hypothetical proteins, given in Supplementary Table S7, also notes the various domains present in these proteins. Some of these proteins may play an important role in the structure and function of glycosomes as these proteins possess domains such as the pentatricopeptide-, tetratricopeptide-, WD-, and chaperone-like domains. Domains such as RING, SNARE, and E3-ligase-like domains indicate that these hypothetical proteins may have a role in the biogenesis of glycosomes. Some of these hypothetical proteins also show homology to proteins that have ribosomal domains, indicating that these are non-glycosomal proteins.
Post-translational modification of proteins
In the current study, we identified 1355 proteins in total from enriched glycosomal fractions. We carried out a comparative analysis with previously published work on post-translational modification (PTM) of proteins in Leishmania parasites (Rosenzweig et al., 2008; Tsigankov et al., 2013; 2014). This comparative analysis of 1355 proteins from enriched glycosomal fractions showed that 266 proteins have a propensity to get post-translational modification. Amongst this phosphorylation was one of the most observed post-translational modification, and 216 proteins are known to be phosphoproteins. The list of phosphoproteins includes enzymes such as phosphoglycerate mutase, enolase, glyceraldehyde 3-phosphate dehydrogenase, phosphoglucomutase, and triose phosphate isomerase, which are involved in carbohydrate metabolism. Post-translational modifications of proteins involved carbohydrate metabolism is in agreement with earlier report by Guther et al., (2014). Similarly proteins involved in redox pathways that were detected in our proteomic study are known to get post-translationally modified. Majority of proteins identified in L. donovani glycosome enriched fraction with post-translational modifications are hypothetical proteins with no known function. We also identified a few proteins that can be glycosylated, acetylated, or methylated when the data were compared with the previous work from Rosenzweig et al., (2008). The complete list of PTM modification identified in glycosomal proteins by comparative analysis is shown in Supplementary Table S8.
Discussion
Leishmania have a number of inimitable characteristics such as the possession of a kinetoplast, a single mitochondrion with a complex kinetoplast-DNA, and the presence of glycosomes. Glycosomes are rudimentary peroxisome-like organelles that contain the major part of the glycolytic pathway. Glycosomes are the metabolic hub within the parasite that help the parasite to survive during differentiation and development within the sandfly gut and host macrophage cells. The parasite needs to survive in different harsh milieu such as exposure to acidic pH and high temperature by adjusting its metabolism to the environment, and glycosomes play an important role in this adaptation.
The proteome of peroxisomes as well as glycosomes varies between species and also between different tissues, developmental stages, and environmental conditions. Proteomic analysis of glycosomes is reported from density gradient purified glycosomes, as well from epitope tagging and immuno-bead pullout techniques (Guther et al., 2014). Here, we have identified 1355 unique proteins in a density gradient fraction enriched in glycosomes by using high resolution mass spectrometry. This profiling has identified proteins that include different enzymes, membrane receptors, and structural proteins. The genome of L. donovani has many genes arranged as tandem repeats that encodes proteins that are paralogs. We have detected many proteins that are encoded by such tandem repeat genes. This is the only study to date to characterize the proteome of L. donovani glycosome-enriched fraction. The study identified some of the important proteins like glycosomal membrane protein, orotidine-5-phosphate decarboxylase/orotate phosphor-ribosyltransferase, transketolase, glucokinase and ribulokinase, 4-coumarate-CoA ligase, arginase, peroxisomal enoyl-coa hydratase, and glycerol-3-phosphate acyl transferase, which were not detected in earlier glycosome proteomic studies.
Only 107 proteins out of 1355 proteins detected in L. donovani promastigote glycosomal proteome were found to have putative PTS motifs which are signals that help in targeting of proteins to glycosomes and peroxisomes. However, it remains to be seen whether these 107 proteins bearing PTS signals are truly glycosomal, since dual localization of PTS bearing peroxisomal proteins is a well-known phenomenon (Ast et al., 2013). Although proteins with the PTS-1 motif can be predicted to be peroxisomal based on their C-terminal tripeptide sequence (Reumann, 2004), the correct prediction of proteins in the peroxisome using an in silico approach based on PTS motifs is known to be an intricate assignment (Chowdhary et al., 2012; Reumann et al., 2012). Although PTS-1 motifs are conserved across the different phyla, in the case of kinetoplastids PTS-1 motifs were reported to be highly variable (Blattner et al., 1992; Sommer et al., 1992).
The search in the Leishmania genome database for PTS-1 sequences yielded unusual motifs that may not be true targeting signals. The PTS-1 signals are classified as strong or weak based on their targeting potential. Weak tripeptide motifs require an auxiliary targeting motif that is present immediately upstream of the PTS-1. This auxiliary motif usually consists of 10 basic amino acids and is essential for targeting proteins to the peroxisomal matrix. These enhancer motifs are ill-defined for most cell types, although have been well characterized for metazoa (Neuberger et al., 2004). As more datasets are being made available for glycosomal proteomic profiles from different kinetoplastid parasites, it will be of interest to study such enhancer elements in kinetoplastids by both experimental and in silico computational approaches.
The added degeneracy of both PTS motifs in Leishmania makes in silico studies more irrelevant until we confirm glycosomal localization by biochemical or immunochemical localization studies. Future studies involving the assessment of the efficacy of different PTS-1 and PTS-2 motifs from L. donovani in targeting reporter proteins, such as green fluorescent protein, to glycosomes will give evidence of the motif's functionality. PTS2-targeting predictions also need to be reviewed with amalgamation of experimental data from targeting studies and receptor and PTS-2 motif binding assays.
Subcellular fractionation and purification of an organelles is known to reduce the complexity in the preparation by essentially removing contaminating proteins, but in this study, we have detected different non-glycosomal proteins in glycosomal fractions. However, it is interesting to note that many of these non-glycosomal proteins are known to play important roles in peroxisome metabolism and proliferation. Such contaminating proteins have been previously reported in different proteomic studies but it was neatly overcome by using SILAC labeling technique while profiling T. brucei glycosomes (Guther et al., 2014). In our previously published proteogenomic study of L. donovani promastigotes and axenic amastigotes, we identified 3999 proteins (Nirujogi et al., 2014). In glycosomal fractions, we could identify 85 proteins that were not detected in the total proteome of the parasite. This indicates that the identification of some low abundance proteins was now possible due to the enrichment of the organelle fraction. Some of these proteins were tryparedoxin peroxidase, dephospho-CoA kinase, UDP-N-acetylglucosamine-dolichylphosphate-N-acetyl-glucosaminephosphotransferase, and phosphatidyltransferase. In addition, this fraction also had membrane proteins such as amino acid transporters, ATP-binding cassette protein subfamily C, flippase, and endosomal integral membrane protein. These 85 proteins also contain hypothetical proteins that have domains which are important in peroxisome and glycosome functions but did include any proteins like the DNA polymerases or topoisomerases. The comparative analysis of all known glycosomal proteome performed in this study also will be useful to identify unique glycosomal proteins for further characterizations.
Conclusions
In our current analysis, we have mapped the proteome of the enriched glycosomal fraction from L. donovani promastigotes. Many of the glycosomal proteins identified in our study are key enzymes in metabolic pathways involved in carbohydrate metabolism, lipid metabolism, thiol metabolism, purine and pyrimidine metabolism. We have also identified many hypothetical proteins; in our proteomic analysis and the roles of these proteins in the Leishmania glycosome are unknown. Various hypothetical proteins detected in this proteomic study have domains suggestive of their role as potential regulators of glycosomal metabolism, which needs to be deciphered. Earlier glycosomal proteomic profiling studies from T. brucei and L. tarentolae and this study from L. donovani will help to identify metabolic enzymes unique to glycosomes, which is crucial for development of new drug targets. Peroxisomes play a vital role in the development and longevity of the multicellular organism, but at the cellular level, peroxisomes are not essential for cell viability and functioning. As opposed to this phenomenon, in unicellular kinetoplastids, mutations in peroxin genes have shown deleterious effects, especially on cell viability. This indicates that unicellular kinetoplastid parasites can be used as an ideal model to study peroxisome biogenesis that may reveal functional aspects of peroxisomes in higher animals. Information from this proteomic profiling will contribute valuable information towards this task.
Footnotes
Acknowledgments
We thank the Department of Biotechnology (DBT), Government of India for research support to the Institute of Bioinformatics. Harsha Gowda is a Wellcome Trust-DBT, India Alliance Early Career Fellow. Harsh Pawar, Gajanan Sathe, and Sandip Chavan are recipients of Senior Research Fellowships from Council of Scientific and Industrial Research, Government of India. Mahendra D. Jamdhade is a recipient of Senior Research Fellowship from the University Grants Commission, Government of India. Kiran N. Mahale is a recipient of post-doctoral fellowship under the DBT-Research Associateship program. We would like to thank Dr. Cheryl Travasso for critical reading and editing of the manuscript.
Author Disclosure Statement
The authors declare that they have no conflicting financial interests.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.