
Abstract
Abstract
Recently developed strategies and techniques that make use of the vast amount of genetic information to perform targeted perturbations in the genome of living organisms are collectively referred to as genome engineering. The wide array of applications made possible by the use of this technology range from agriculture to healthcare. This, along with the applications involving basic biological research, has made it a very dynamic and active field of research. This review focuses on the CRISPR system from its discovery and role in bacterial adaptive immunity to the most recent developments, and its possible applications in agriculture and modern medicine.
Introduction
T
Genome engineering is a considerably broader term that encompasses technologies to make targeted modifications to the genome, its contexts (epigenetic marks) and function (transcripts) (Hsu et al., 2014). Besides the applications in functional genomics and understanding organization at a systems level, this technology provides a platform to make targeted mutagenesis and transgene insertion for gene therapy and for crop and livestock improvement. Thus, genome engineering enables the cell's DNA to be precisely manipulated for a wide array of applications in the fields of basic biology, agriculture, medicine, and biotechnology.
Genome editing is performed by triggering the cell's DNA repair mechanisms. Development of gene targeting through homologous recombination (HR) was the first major breakthrough in genome modification. This mechanism integrates exogenous genes into the genome of a cell based on homology between target sites and donor sequences and has been used to create knock-out and knock-in mice with the germ line stem cells, although the frequency of recombination was very low (1 in 106–9) (Capecchi, 1989). Gene targeting for knockout, modification, or replacement of genes have been performed in a number of plants and is reviewed extensively (Chen and Gao, 2014; Lozano-Juste and Cutler, 2014; Voytas, 2013; Voytas and Gao, 2014).
Researchers over 20 years ago demonstrated that induction of double-strand breaks (DSBs) in the genome at target sites could stimulate genome editing through HR mediated repair (Rudin et al., 1989). Programmable sequence-specific nucleases can be used to introduce these DSBs, leading to modification at cleavage sites that can be directed or random (Bibikova et al., 2001). Cells have two major repair pathways in response to DSB: non-homologous end joining (NHEJ) and homology directed repair (HDR) pathways. The HDR repair pathway is activated if a designed donor template is available at the DSB. This mediates precise substitution of the donor template at the DSB site replacing the earlier sequence (Bibikova et al., 2001, 2003; Rudin et al., 1989). In the absence of template at the site, the DSBs are repaired through the NHEJ which is error prone, mostly resulting in addition or deletion of nucleotides at the site of repair (Bibikova et al., 2001, 2003; Rudin et al., 1989).
Different genome-editing systems have been developed rapidly in the past few years. CRISPR-Cas9 technology is based on the RNA guided endonuclease (RGE) Cas9, which can be used to target specific location of choice in the genome of virtually any organism with the help of a short RNA guide. A number of good reviews have already been published describing the CRISPR-Cas9 system, its history, and development into a powerful molecular tool for genome-engineering applications in the fields of applied and basic biology (Doudna and Charpentier, 2014; Hsu et al., 2014; Mali et al., 2013b; Voytas and Gao, 2014). Here we will elaborate the scope, history, mechanism, protocols, bioinformatic tools, applications, and prospects of the CRISPR-Cas9 genome engineering technology in all aspects of biological, agricultural, and biomedical research.
Genome Perturbation: Engineered to Programmable Nucleases
As mentioned earlier, DSBs trigger either the NHEJ repair resulting in indels (Bibikova et al., 2002) or HDR directing precise template-dependant substitution at the DSB (Bibikova et al., 2001, 2003; Hsu et al., 2014; Rudin et al., 1989). Research in this field is focused on engineering sequence-specific nucleases (SSNs) to cleave at target locations in the genome, generating DSBs that can be manipulated using these repair pathways. The NHEJ triggered indels are useful for generating targeted gene knockouts, whereas the HDR-based repair mechanism can be used to insert a gene, fix or replace a mutated allele for performing targeted genetic modification or gene therapy. Thus DSB induced HDR and NHEJ pathways are established as precise mechanisms of targeted genome editing in eukaryotes.
So far, four types of engineered SSNs have been used for targeted genetic alternations (Fig. 1).
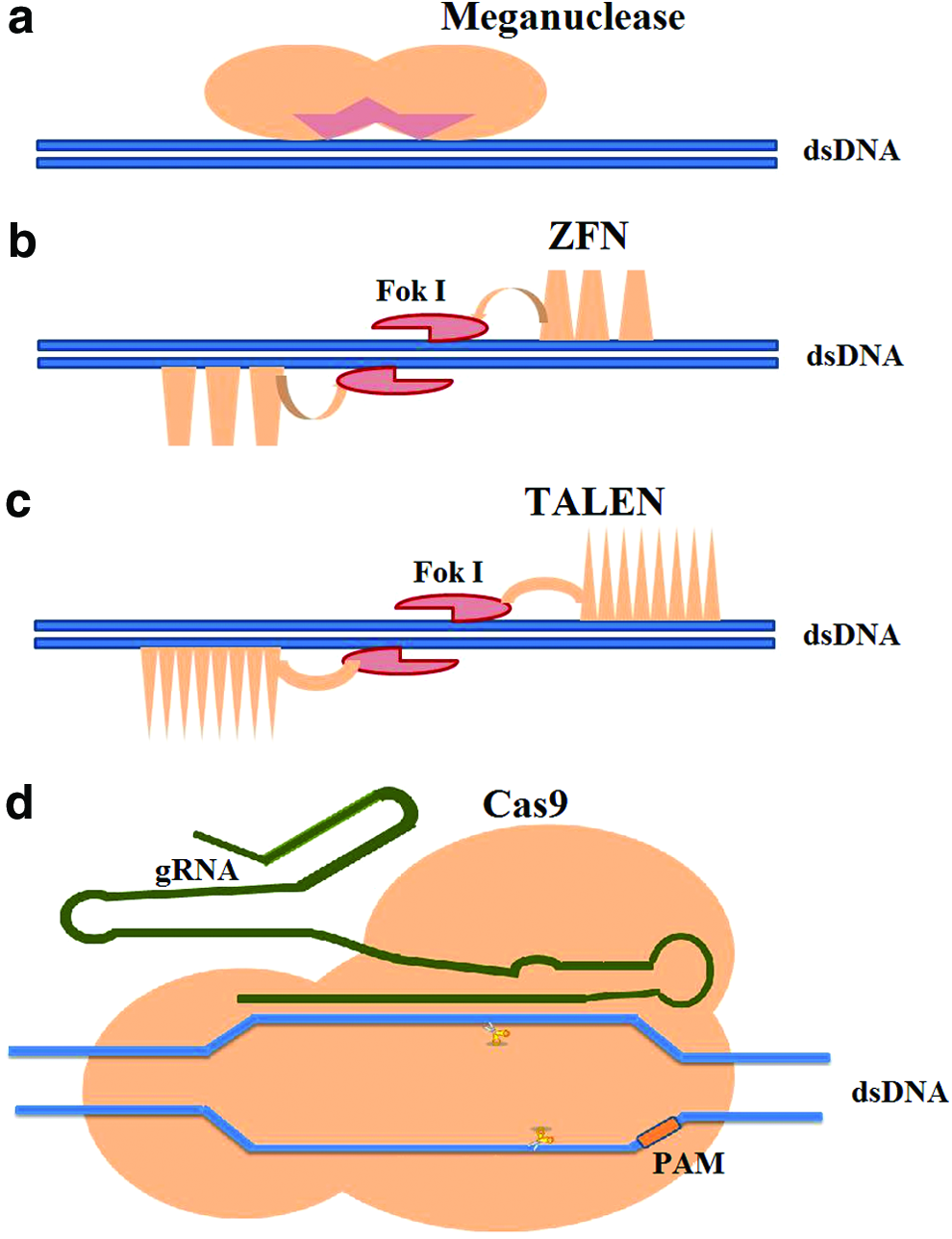
The four major categories of sequence specific nucleases: (a) Meganucleases have the base recognition and nuclease domains fused together; (b) Zinc finger nucleases acting as a dimer for making DSB, a monomer consists of three zinc fingers, each making base specific interactions. The Fok I nuclease domain is tethered to the array; (c) TALE array consists of repeat variable di-residues that can make base specific contact with the target DNA sequence. It also has Fok I endonuclease domain fused to it. (d) The Cas9 endonuclease is guided by the chimeric gRNA to target sequence based on complementary base pairing and the appropriate PAM sequence. Adapted from previous publications (Mali et al., 2013; Voytas, 2013).
Among the four systems mentioned above, Meganucleases, ZFN, and TALENs function through specific DNA–protein interactions, while CRISPR-Cas9 action is mediated through DNA–RNA–protein interaction. Re-engineering the meganuclease DNA recognition domain has proven to be more difficult compared to those of ZFN and TALENs because it has integrated DNA binding and nuclease domains, which lack clarity in terms of causal relationship between the amino acid sequence and target DNA specificity (Pauwels et al., 2014). In ZFN that functions as a dimer, DNA binding is facilitated by an array of zinc fingers in which each individual module specifies a 3-nt target (Voytas, 2013). In contrast, the TALEN specificity is provided by amino acid residues acting in pairs, each of which binds to a specific nucleotide (Moscou and Bogdanove, 2009). TALENs being highly modular are easily designed as compared to ZFN. Systems utilizing ZFN suffers from context-dependent specificity (Carlson et al., 2012) (i.e., the design of ZFN must take into consideration various parameters such as the flanking region and epigenetic state of the DNA). Both ZFN and TALEN technologies demand elaborate design, assembly, and screening of each individual DNA-binding protein for a particular target site (Hsu et al., 2014).
TALEN and ZFN are both designer nucleases requiring re-engineering for each different intended target (Carlson et al., 2012). However, in CRISPR-Cas9 system, the Cas9 enzymes can be specified to an array of targets simply using a group of guide RNA (gRNA), which makes the use of CRISPR-Cas9 system easier. In this strategy, only the 20-nt targeting sequence lying within the guide RNA needs to be changed in order to target different genes. This makes the RNA-guided CRISPR-Cas9 system relatively robust, affordable, and easiest to engineer among the four designer nucleases. The short guide RNA recognizes the target sequence via Watson-Crick base pairing.
In prokaryotes, the CRISPR-Cas9 system provides a natural defense mechanism against bacteriophages by using the guide RNAs to direct RNA-dependent Cas9 to corresponding phage sequences that are targeted and cleaved. In order to reprogram Cas9 enzyme to target a specified location in the genome, a sequence of interest can be used to replace the phage sequence in the guide RNA. In this way, Cas9 can be used as a guided missile to target any location in the genome through gRNA. Instead of using a number of bulky engineered proteins such as TALENs or ZFNs for simultaneous targeting of multiple sites, a library of gRNA along with a single Cas9 can efficiently perform multiplex genome editing (Cong et al., 2013). A study conducted in maize showed that TALEN and Cas9 have similar mutagenesis efficiency for intended target sites (Liang et al., 2014). Cas9 and TALEN are also shown to have comparable efficiency for HDR-directed modifications in the Drosophila genome (Yu et al., 2014).
Development of the CRISPR-Cas9 System
The CRISPR-Cas9 story began in 1987 with the discovery of a mysterious locus in the E. coli genome adjacent to the iap gene (Ishino et al., 1987). It was described as a series of direct repeating DNA sequences (29 nt) intervened by similar sized, nonrepetitive DNA (32 nt). Further studies and in silico analysis identified these motifs (27–37 nt) in many sequenced bacteria (40%) and archaea (90%) (Mojica et al., 2000). These repetitive elements were initially called short regularly spaced repeats (SRSR), but were later changed to clustered regularly interspersed short palindromic repeats (CRISPR) (Jansen et al., 2002; Mojica et al., 2000). Interspersed sequences within the repeats called ‘spacers' are generally of similar size. A search for the identities of these CRISPR spacers led researchers to know about their homology to extra-chromosomal (conjugative plasmids) and bacteriophage DNA, which is known to be related to spacer-containing prokaryote (Bolotin et al., 2005; Mojica et al., 2005; Pourcel et al., 2005).
The phage-resistant bacterial strains expressed the CRISPR loci (Tang et al., 2002), suggesting its involvement in prokaryotic adaptive immune memory and response (Mojica et al., 2005). It was observed that phage-resistant strains became susceptible upon modification of corresponding spacer, and gained resistance upon addition of novel spacers to the CRISPR loci (Barrangou et al., 2007; Datsenko et al., 2012). Genes flanking CRISPRs were identified and shown to be conserved, and were named CRISPR-associated (cas) genes (Jansen et al., 2002). Computational analysis demonstrated genes encoding Cas proteins have nuclease, helicase, polymerase, and various DNA and RNA binding domains (Jansen et al., 2002). While cas genes are translated into proteins, the CRISPR sequences are transcribed as long RNA molecules that are later processed into shorter CRISPR RNA (crRNA).
The mechanism of prokaryotic CRISPR-based acquired immunity has been described in three steps: (1) spacer acquisition, (2) crRNA processing, and (3) interference (Makarova et al., 2006). In the first step, spacer integration into CRISPR locus is mediated by Cas1-Cas2 complex (Nuñez et al., 2014). In the incoming foreign phage, Cas1 recognizes a unique protospacer adjacent motif (PAM), near to which a sequence of defined length (protospacer) is excised by it (Makarova et al., 2011b). The excised sequence, along with a single nucleotide of PAM, is integrated into the CRISPR locus of the host genome and completes the acquisition (Swarts et al., 2012).
Based on machinery of crRNA processing and interference that are variable in prokaryotes, there are three types of CRISPR systems (reviewed in Chylinski et al., 2014; Makarova et al., 2011b). crRNA processing and interference in type I and type III CRISPR systems have been shown to involve multi-ribonucleoprotein complexes such as CASCADE proteins (type I) and RAMP module (Type III) (Brouns 2009; Hale et al., 2013). In comparison, type II CRISPR-Cas system is less complex, making it suitable for development into a powerful genome-editing tool. During the second step, pre-crRNA processing is guided by the binding of trans-encoded small RNA (tracrRNA) (Deltcheva et al., 2011), which directs RNase III catalysis, leading to cleavage of hybridized pre-crRNA-tracrRNA in the presence of Cas9. The cleaved mature crRNA remains associated with tracrRNA and Cas9 (Deltcheva et al., 2011; Xing et al., 2001). The spacer sequence in the crRNA makes complementary base pairing with the target sequence and guides Cas9 (previously named as Cas5, Csn1 or Csx12), the nuclease enzyme in type II CRISPR, (Garneau et al., 2010). The tracrRNA also hybridizes to crRNA and plays an important role in facilitating RNA-guided targeting of Cas9 (Deltcheva et al., 2011).
The target-cleaving mechanism must also distinguish itself (CRISPR locus in the genome) from non-self (i.e., invading phage). This is possible by PAM, which is required along with protospacer for target cleavage (Gasiunas et al., 2012). PAM acts as a targeting component that differentiates self from non-self and prevents CRISPR from targeting its own locus. PAM also dictates the target search mechanism of the Cas9 enzyme that is elaborated in the later sections. Following its discovery and characterization, there arose the possibility of developing the RNA-guided interference mechanism of Cas9 into a programmable genome editing tool. Various groups also started to use the natural CRISPR array for production of phage-resistant microbial cultures for dairy industry (Quiberoni et al., 2010).
Functional Organization, Diversity, and Activity of Cas9
The structure and diversity of the CRISPR-Cas9 system have already been extensively reviewed (Chylinski et al., 2014; Hsu et al., 2014; Mali et al., 2013b). The Cas9 enzyme has a recognition (REC) and nuclease (NUC) lobe along with a PAM interacting (PI) C-terminal domain (CTD) (Nishimasu et al., 2014). The NUC lobe consists of two nuclease subdomains, RuvC and HNH. The noncomplimentary and complimentary strand of the target are bound and cleaved by RuvC and HNH subdomains, respectively, creating a DSB in the DNA during ‘interference’. If either of the nuclease domains is dysfunctional, the Cas9 enzymes produce targeted nicks instead of DSBs and such enzymes are referred as nickases (Mali et al., 2013a). The α-helix rich REC lobe recognizes and facilitates target binding.
Basically, the Cas9 structure folds and re-organizes itself around the gRNA in a manner that facilitates target DNA binding (Nishimasu et al., 2014). It is inferred that Cas9 activity is triggered upon gRNA binding to target DNA forming the RNA–DNA duplex. When Cas9 does not bind to gRNA and target DNA, it remains in an auto-inhibited conformation (Jinek et al., 2014). It is understood that certain defined regions of the gRNA are necessary for its transferability as well as activity of Cas9 in different systems (Briner et al., 2014). PI-CTD binding to PAM is an absolute requirement for enzyme function. PI-CTD confers PAM specificity and also governs Cas9 activity through steric hindrance (Nishimasu et al., 2014). PAM recognition by the PI-CTD is a prerequisite for ATP-independent-strand-separation of target DNA, followed by DNA (target)–gRNA heteroduplex formation (Anders et al., 2014).
Homology-based searches in sequence databases have identified more than thousand Cas9 nucleases from different bacteria (Chylinski et al., 2014; Makarova et al., 2011b). All of these are exclusively associated with the type II CRISPR system and are further classified as subtypes (IIA–IIC) based on the structural organization of the CRISPR locus (Makarova et al., 2011a). Cas9 proteins can also be categorized into groups on the basis of size (1100, 1350, or1500 amino acids). Despite all the variations in length, all Cas9 proteins have the same domain architecture and organization [NUC (RuvC and HNH domains) and REC nodes along with the PI-CTD]. Variations in size lie within the REC node that is responsible for the association with gRNA and target sequence (Jinek et al., 2014). Sequence coding for REC node is poorly conserved and it can be easily modified by means of recombination and truncation. Such modifications on the REC node holds promise for re-engineering Cas9 for optimizing parameters such as DNA binding, cleavage and protein size (Hsu et al., 2014). Unlike the Type I and Type III CRISPR systems which are found in both bacteria and archeae, Type II CRISPR system is only found in bacteria (Chylinski et al., 2014; Hsu et al., 2014).
Cas9 associates with gRNA or the crRNA-tracrRNA hybrid forming a ribonucleoprotein complex that initiates the target search mechanism. The interaction between PI-CTD of Cas9 and PAM sequence at the 3’ flanking region of the target site facilitates strand separation via the action of phosphate lock loop of the CTD stabilizing unwound DNA (Anders et al., 2014). This indicates that Cas9-gRNA complexes initially associate with the PAM sequence, thereby initiating separation of the target strands for the binding mechanism. PAM recognition followed by gRNA complementarily binding to the target sequence creates conformational changes in HNH and RuvC domains and activates nuclease activity (Nishimasu et al., 2014).
CRISPR-Cas9: Advancement as a Versatile Genome Editing Tool
Successful interference against exogenous plasmid and phage infection by CRISPR II system following transfer from S. thermophilus to E. coli effectively demonstrated the transferability of Cas9 system for the first time (Sapranauskas et al., 2011). Also, purified Cas9 (derived from S. thermophilus and S. pyogenes) has been shown to target desired DNA sequence in vitro (Gasiunas et al., 2012; Jinek et al., 2012). The SpCas9 derived from S. pyogenes cuts 3 bp upstream of the PAM making a blunt DSB (Jinek et al., 2012). As mentioned earlier, Cas9 activity is dependent on crRNA to specify the target sequence and tracrRNA to facilitate the cleavage. To reduce complexity, a chimeric short guide RNA (sgRNA or simply gRNA) is constructed by fusing tracrRNA and crRNA. This chimeric gRNA has been shown to facilitate DNA cleavage in vitro (Jinek et al., 2012).
Genome editing was successfully demonstrated in mammalian cells using Cas9 from S. thermophilus and S. pyogenes with appropriate gRNA (Cong et al., 2013; Mali et al., 2013b). The gRNA or crRNA-tracrRNA hybrid directed Cas9 cleavage in the mammalian cell genome to stimulate either NHEJ or HDR-based editing. Cas9 (SpCas9) has already been used for genome editing in a wide variety of organisms and cell types ranging from bacteria, yeast, zebrafish, roundworm, fruitfly, crop plants, mouse, goat, monkey, to human cell lines (Sander and Joung, 2014). The number of experimental organisms in which Cas9-mediated genome editing has been demonstrated is ever increasing.
The CRISPR system in prokaryotes encodes multiple spacers in crRNA that are processed to act as guides individually specifying cleavage of each target. The ability to individually specify target cleavage is the major advantage of the CRISPR system in genome editing. The Cas9 enzyme can be incorporated along with a library of gRNAs to target multiple locations in the genome. This process is referred to as multiplexing and it has been successfully demonstrated in mammalian and plant [Bread wheat (Triticum aestivum)] cells with an array of spacers in a battery of gRNA (Cong et al., 2013; Mali et al., 2013b; Wang et al., 2014). However, it is seen that multiple gRNA stacked in expression cassettes exceeds size limitations for cloning, thereby lowering the overall efficiency. A novel strategy to overcome this problem involves repeats of gRNA and tRNA integrated into a synthetic gene. The expressed transcript is recognized by tRNA processing enzymes which cleave at the junctions, precisely releasing the multiple gRNAs that is targeted simultaneously to multiple sites. The universality of the tRNA processing enzymes in life forms enables this multiplex genome engineering technology to be applied to virtually any organism (Xie et al., 2015).
Target Range and Specificity of Cas9
The PAM sequence recognition determines a lot of parameters of the Cas9 activity; these include, self vs. nonself determination, target search mechanism, target strand separation, and the transition between target binding and cleavage conformations (Anders et al., 2014; Gasiunas et al., 2012; Hsu et al., 2013; Nishimasu et al., 2014). In addition to these indispensable functions, PAM sequences are also important in determining targeting space of Cas9. For example, SpCas9, whose PAM sequence is NGG, allows it to target roughly every 8 bp within the human genome (Cong et al., 2013; Hsu et al., 2013, 2014). The PAM sequence is specific to a particular Cas9 ortholog, as in the case of S. thermophilus, which has two Cas9 homologs, each having a different PAM sequence specificity (Hsu et al., 2014). Cas9 equipped with additional PAM sequences will expand targeting range of the technology. Such additions of new Cas9 homologs are possible either through computer-aided metagenomic analysis of prokaryotes (Chylinski et al., 2014) or by manipulating established PI domains to recognize new PAM sequences (Nishimasu et al., 2014). By manipulating PI-CTD, one can reduce and modify PAM size to improve flexibility of Cas9 for PAM recognition. Orthogonal genome engineering is benefited by the delivery Cas9 proteins with different PAM specificity.
Simultaneous functional manipulations to various locations within the same host genome are possible with the help of different Cas9 enzymes with varying PAM specificity. For example, Cas9 enzymes with independent nuclease and transcriptional repression, actively function inside a host simultaneously performing different functions in different loci (Esvelt et al., 2013). Cas9 activity in terms of chromatin accessibility is still not well defined at this point. The functionality of Cas9 in the heterochromatic regions needs validation. dCas9, the nuclease inactivated form of Cas9, functions as a fusion protein with effectors such as transcriptional activators tethered to it. In fact, dCas9 (Cas9 nuclease deficient) fusion protein has been shown to induce expression from loci found within the inaccessible chromatin (Hsu et al., 2014; Perez-Pinera et al., 2013). DNA methylation states extend negative influence on TALEN activity (Valton et al., 2012). However, DNA cleavage by Cas9 either in vitro or in vivo is unaffected by methylation (Hsu et al., 2013, 2014).
Off-target cleavage is a major cause of concern especially in health applications since the modifications made are permanent. Therefore, Cas9 specificity has been extensively tested and analyzed by using a library of mismatched gRNA, in vitro selection, and reporter assays (Fu et al., 2013; Hsu et al., 2013; Pattanayak et al., 2013). The study headed by Joung and Sander demonstrated that SpCas9 can tolerate up to five mismatches between gRNA and target DNA for mutagenesis in an off-target site (Fu et al., 2013). It is easy to evaluate the influence of mismatches in gRNA on Cas9 activity because the latter is dependent on complementarity with target DNA. This evaluation is not possible with ZFNs and TALENs because of the difficulties involved in preparing DNA binding protein libraries having variable specificity for target sequences.
Cas9 specificity studies illustrate that mismatch tolerance depends on the number, position, and distribution of the mismatches (Hsu et al., 2013, 2014; Mali et al., 2013a). Researchers infer mismatches closer to PAM sequence are more significant, and the mismatches farther from PAM have lower impact on Cas9 specificity. It has been observed that Cas9 temporarily binds to several off-target sites in the genome without cleaving them (Wu et al., 2014). PAM recognition triggers transient Cas9 binding, but extensive interaction between PAM distal sequences and the 5’ end of gRNA is necessary for mediating cleavage (Cencic et al., 2014). Concerns about off-target activity vary depending on application of Cas9 and tend to be less when Cas9 is intended as a sequence specific nuclease.
Higher levels of concerns arise when Cas9 is used as a targeted DNA binding device, also referred as programmable homing device, which sometimes binds at several unspecified sites throughout the genome. Enzymatic concentration is also an important parameter for off-target activity of Cas9 with more errors seen at higher concentrations of enzyme. Studies by Hsu et al. (2013) demonstrated that the specificity increased drastically when the concentration of sgRNA and Cas9 used in transfection was reduced from 400 to 10 ng of Cas9-gRNA plasmid. Similarly, four-fold and seven-fold increase in specificity was observed when amounts of plasmid transfected were reduced from 400 ng to 50 ng and 50 ng to 10 ng, respectively.
Three strategies have been devised to improve targeting fidelity of Cas9. The first strategy is to employ nickases making targeted single stranded breaks in DNA under the direction of suitable gRNA. As mentioned earlier, inactivating either of the nuclease domains (HNH or RuvC) results in Cas9 becoming a nickase, which makes nicks instead of DSB (Gasiunas et al., 2012; Hsu et al., 2014; Jinek et al., 2012; Nishimasu et al., 2014). Properly spaced nicks in the genome can be created by Cas9 nickase specified with a pair of gRNA. Such appropriately targeted nicks can duplicate the effects of DSB (Mali et al., 2013a; Ran et al., 2013). Off-target nicks are repaired efficiently by the base excision repair machinery in the cell, as these will not have the associated, cooperative nick (Dianov and Hübscher 2013; Hsu et al., 2014). However, further investigations are needed to compare Cas9 nickases and wild-type Cas9 in the activation of HDR for precise substitution of sequences.
The second strategy to increase target specificity of Cas9 is by the use of truncated gRNA (Fu et al., 2014). Binding affinity is lowered when mismatches exist between target and gRNA, but these are mostly tolerated during off-target cleavage of Cas9 due to excessive binding energy. Truncated gRNA limits excessive binding affinity with target DNA and reduces Cas9 enzyme's mismatch tolerance. These strategies can be combined to reduce off-target editing (Fu et al., 2014).
The third strategy developed to improve cleavage specificity is the use of fCas9, which was generated by fusing the Fok I nuclease domain to a catalytically inactive Cas9 (Guilinger et al.2014). Two fCas9 monomers are needed to make a DSB, analogous to the functioning of nickases. Results of this study showed fCas9 has a four-fold higher specificity than paired nickases at loci with similar off-target sites and >140-fold higher specificity than wild-type Cas9. The gRNA modules necessary for Cas9 activity and orthogonality have been identified recently (Briner et al., 2014), and these findings promote better design of gRNA. Several tools for target selection and gRNA design along with prediction of off-target sites are already available for a wide range of organisms, and these are described in Table 1. The nonprofit plasmid repository, Addgene, facilitates exchange of CRISPR plasmids and protocols between labs for scientific and medical research applications (http://www.addgene.org/CRISPR).
Genome Engineering: Expanding the Usage of CRISPR System
Wild-type Cas9 or Cas9 nickases can be used to make multiple targeted genome modifications in several organisms. They can be orthogonally multiplexed to probe gene function (loss-of-function screen) and genetic variations in a large scale (Hsu et al., 2014). Cas9 also mediates knock-in of genes via the HDR pathway as mentioned earlier. Recently an alternative strategy (CRIS-PITCh) was introduced to knock-in genes based on microhomology-mediated-end-joining (MMEJ) (Nakade et al., 2014), which circumvents the problems associated with organisms or cells having low HR efficiency. Another strategy involves the use of small molecules to positively modulate HDR efficiency (three-fold) of Cas9 system (Yu et al., 2015). Cas9 can be developed into a targeted homing device (dCas9) on the genome by inactivating its nuclease domains; this technology has been named CRISPRi (see below). In addition, the applications of effectors fused with dCas9 expanded Cas9 use by enabling it to perform a wide range of genome-engineering applications. Individual effectors such as transcriptional activators, repressors, and chromatin remodelers can be selectively recruited to a desired location in the genome by attaching them with the homing device-dCas9 (Mali et al., 2013b). Applying different modified forms of Cas9 expands possibilities of genome engineering that can be defined as targeted perturbations to manipulate the structure (sequence), context (epigenetics), and function of the genome. Below are extended applications made possible through the modified Cas9:
Transcriptional modulation
In CRISPRi, the dCas9 binds to a particular location in the genome and represses transcription by hindering RNA polymerase activity. This approach is particularly useful and effective for repressing expression in prokaryotes, but has been less effective in eukaryotes (Gilbert et al., 2013; Hsu et al., 2014). In eukaryotes, this is enforced by tethering dCas9 with repressor domains such as SID4X and KRAB, which silence genomic loci through chromatin modification (Gilbert et al., 2013; Konermann et al., 2013). Similarly, transcriptional activator domains VP16/VP64 derived from viral trans-acting proteins, when fused to dCas9, have been shown to enhance the expression of targeted genomic loci (Hsu et al., 2014; Konermann et al., 2013; Mali et al., 2013b). However, it is necessary to utilize several gRNA coinciding to a single genomic locus to achieve effective transcriptional activation. This process of targeting a single locus with multiple gRNA is called tiling (Maeder et al., 2013; Perez-Pinera et al., 2013).
Based on the crystal structure of the Cas9–gRNA–DNA tertiary complex (Nishimasu et al., 2014), a recent study demonstrated gRNA stem loop 2 and tetraloop tolerates the addition of protein interacting aptamers. It provides a better anchoring position for the activation domain which is recruited via aptamers, enhancing expression (12-fold) compared to dCas9-VP64 (Konermann et al., 2014). Light-induced transcriptional activation was demonstrated in HEK293T cells by tethering light inducible heterodimerizing proteins to dCas9 (Light-activated Cas9 effector (LACE)) (Polstein and Gersbach, 2015). Inducible transcriptional activation and genome editing are accomplished using split-Cas9 fragments. These fragments are rendered active via rapamycin binding dimerization domains, which enable controlled reassembly of Cas9 to mediate transcriptional modulation and genome editing (Zetsche et al., 2015).
Epigenetic modification
The epigenetic modifications on the genome tune the state of differentiation and govern the cell fate (Victor et al., 2014). Transcription activator like effectors (TALE) bound with TET1 can induce demethylation at the CpG sites, which is also predicted to be possible with dCas9 to activate genes repressed by CpG methylation (Eguchi et al., 2014; Konermann et al., 2013). Potentially, artificial epigenetic modulators (Cas9, TALE, and ZF) can function independently of cellular states and signals, making them promising tools for modifying cell fate (Eguchi et al., 2014). Orthogonal genome engineering is functionally variant manipulation of the genome simultaneously at different loci within the same cell or in cell population. Orthogonal epigenetic and transcriptional regulatory systems for simultaneously activating, repressing, or modifying specific loci in the genome could be made by using multiple Cas9 proteins of varying PAM requirements (Hsu et al., 2014). Potential crosstalk between endogenous regulatory elements and effector domains needs to be assessed. Hsu et al. (2014) suggested the application of prokaryotic epigenetic enzymes to minimize such crosstalk.
enChIP and imaging
Specific sequences of DNA can be purified with the help of dCas9 used with an epitope or affinity-based tag. This purification process is called enChIP (engineered Chromatin immunoprecipitation) and when used in combination with mass spectrometry (enChIP-MS) can be used to identify proteins associated with genomic loci (Fujita and Fujii, 2013). This method has been employed to identify telomere-binding molecules with engineered TALE (Fujita et al., 2013). dCas9 as a homing device when fused with a fluorescent marker like GFP can also be used in imaging. However, tiling must be done to obtain a detectable signal. Techniques such as in situ hybridization require chemical fixation and cannot be performed in live cells, whereas live cell imaging is possible with dCas9 (Chen et al., 2013; Hsu et al., 2014). Additionally, orthogonal Cas9 proteins tagged with different colored reporters along with multiple gRNA can perform multicolor and multi-locus tagging for nuclear localization and chromosomal architectural studies (Hsu et al., 2014).
Manipulation of ssDNA and ssRNA
Cas9 has been shown to target, modify, and substitute single-stranded oligodeoxyribonuceotides (ssoDNA) (Hwang et al., 2013). In addition, a recent study conducted in the Doudna lab demonstrated successful use of RCas9 to cleave ssRNA (O'Connell et al., 2014). The ability of RCas9 to modify RNA has the potential to transform the study of RNA function through direct (tag-less) detection, analysis, and manipulation of transcripts. This RCas9 system consists of a programmable gRNA-guided Cas9 along with complimentary PAMmer (PAM presenting oligonucleotides), RCas9 can recognize and bind specific ssRNA corresponding to the gRNA (O'Connell et al., 2014).
Thus, Cas9 and its modified versions can dramatically increase the array of tools available for genome engineering. It is an easy, reprogrammable and versatile tool for engineering living organisms by editing the genome sequences, altering transcriptional states of genomic loci, changing chromatin states, and rearranging 3D organization of the genome (Hsu et al., 2014).
Delivery and Expression System for CRISPR-Cas9
Generally, the CRISPR-Cas9 system is delivered to mammalian cells via direct plasmid transfection (Long et al., 2014; Wang et al., 2013). In plants, PEG-mediated protoplast transformation or Agrobacterium-mediated transformation has been employed (Jiang et al., 2013). In recent years, vectors derived from retrovirus and lentiviruses (a complex form of retrovirus) have become essential tools for delivering nucleic acid to different cell types. Mechanisms involve the reverse transcriptase, providing the ability to retrotranscibe their RNA genome into cDNA, which viruses insert into host genomes (Daya and Berns, 2008). Lack of pathogenicity and ability to infect both dividing and nondividing cells also make Adeno-associated virus (AAV) a safe and frequently used viral vector for gene transfer, especially in animal models (Daya and Berns, 2008). Non-integrating viral vectors are also available for transient delivery and expression (Bobis-Wozowicz et al., 2014).
A disadvantage of commonly used lentiviral and AAV vectors is an insufficient capacity to contain the entire Cas9 system (Cas9 with gRNA and other elements such as promoters, reporters, and polyadenylation sequences) for delivery (Ding et al., 2014; Wu et al., 2010). Viral vectors with higher-carrying capacity, such as the adenovirus also have disadvantages in terms of higher immunogenicity and limited cell-type specificity (Ding et al., 2014). Cas9 and gRNA expression cassettes in separate plasmids are generally co-transfected, although larger capacity pX330 vectors (from Feng Zhang lab) (Cong et al., 2013) can be used to accommodate both gRNA and Cas9-encoding sequences simultaneously. The all-in-one vector system containing six gRNA with a nickase-encoding sequence, constructed using Golden Gate cloning method into a single vector, has been successfully employed for multiplex genome editing in HEK293T cells (Sakuma et al., 2014). In order to overcome the delivery challenges associated with the large Cas9 protein (>4 kb) and for efficiently performing in vivo modeling, Cas9 knock-in mice expressing Cas9 in a Cre-driven manner, has been generated (Platt et al., 2014). It is available as a resource for in vivo and in vitro genome editing which is specified by the gRNA delivered (Platt et al., 2014).
Hydrodynamic injection has also been employed for in vivo genome editing in mouse liver (Platt et al., 2014; Yin et al., 2014). Lentiviral vector delivery is hindered by template switch-mediated recombination occurring between the repetitive elements during reverse transcription in case of TALEN delivery. To improve the efficient transfer of mRNA using lentiviral vectors, Mock et al. (2014) proceeded with an approach to disrupt the reverse transcriptase. In doing so, they demonstrated transient expression of SSN in mammalian cells through delivery of its mRNA through lentiviral vectors with modified RT (Mock et al., 2014). In order to realize the potential of protein-based therapeutics, intracellular delivery of proteins is needed (Zuris et al., 2014). Recently, a novel method has been developed to directly deliver proteins involved in genome engineering (Cre, Cas9, dCas9, and TALEN) into cells. These proteins involved in genome engineering are fused to highly anionic supercharged GFP, which are delivered using cationic liposomes to human cell lines and mice enabling efficient genome engineering (Zuris et al., 2014). Timed delivery of the Cas9-gRNA ribonucleoprotein complexes in HEK293T cells also resulted in higher efficiency HDR (Lin et al., 2014).
In plants, Voytas and colleagues reported the use of Gemini Virus Replicons (GVR) for the amenable delivery and expression of CRISPR-Cas system or TALEN for genome engineering. GVR can be combined with other methods for delivery of sequence specific nucleases, such as T-DNA (Baltes et al., 2014; Voytas and Gao, 2014). Unlike Agrobacterium-mediated transformation, Gemini virus can infect monocots and dicots, making it highly versatile as a vector and suitable for engineering many crops (Mach, 2014). The CRISPR-Cas system (along with a donor template for HDR) can be co-delivered into plant cells transiently either as DNA through viral vectors, Agrobacterium, particle bombardment (Baltes et al., 2014) or as mRNA and proteins (Voytas and Gao, 2014). Ideally, SSN are transiently introduced to carry out the desired inherited change in the genome without becoming integrated into the host genome and are degraded eventually. Even if integration occurs, the construct can be segregated from the genome by crossing to obtain a transgene free modification in the plant (Voytas and Gao, 2014).
Genome Engineering: Developments to Possibilities in Agriculture
The potential of the CRISPR-Cas system in crop improvement has yet to be realized in its entirety. Several proof of concept studies have demonstrated the possibility of genome editing in plants, and most of the published studies describe gene knockouts in various plants. This approach of making knockouts is essential for elucidating gene function. The Cas9 system is especially useful as a tool to knock out the diverse array of redundant genes and gene families in plants that resulted from gene duplication and polyploidization (Voytas and Gao, 2014). Cas9 can be harnessed to target individual genes or multiplexed for targeting several gene family members. This versatility enables study of the genetic basis of many previously untraceable and complex traits in plants. A handful of model plants and crops have been targeted with the CRISPR system. These include Arabidopsis (Li et al., 2013), tobacco (Li et al., 2013), rice (Shan et al., 2013), wheat (Wang et al., 2014), sorghum (Jiang et al., 2013b), maize (Liang et al., 2014), tomato (Brooks et al., 2014), and sweet orange (Jia and Wang, 2014) (Table 2).
Gene knockouts have significant impact on new trait development, as can be seen from the vast array of commercialized natural and induced mutant genotypes that have changed the agricultural landscape as a whole (Ahloowalia et al., 2004). Application of SSN for targeted gene knockout is a great improvement over the random process of chemically induced nonspecific mutations. Before SSN, RNAi technology has long been employed to knock down targeted genes in a spatial and temporal manner to study gene function. However, sustained expression of the RNAi construct is needed for disrupting gene function in a particular tissue, making it undesirable in the agriculture setting (Voytas and Gao, 2014). Another advantage of SSN over RNAi technology is that it generates null allele while low level of transcripts of the gene-of-interest still can be present in case of RNAi, which can confound the interpretation of data.
The CRISPR-Cas9 system offers several advantages over RNAi, because it targets duplicated genes in the genome, modifying several alleles. For example, powdery mildew disease is one of the most destructive wheat diseases. It causes up to 40% damage during severe infection with optimal environmental conditions (Gil-Humanes and Voytas, 2014). Powdery mildew is caused by the fungus Blumeria graminis f. sp. tritici. Recently, the Gao–Qiu groups have used Cas9 technology to target and mutate three mildew-resistance locus (MLO) homoeoalleles in bread wheat (Triticum aestivum L.), a hexaploid crop (Wang et al., 2014). Self-fertilized T0 plants resulted in progeny with all the six alleles knocked out, which conferred resistance to the powdery mildew disease. Broad spectrum resistance to powdery mildew can be incorporated into both monocot and dicot crops through this approach, which otherwise depends on heavy use of fungicides (Gil-Humanes and Voytas, 2014). A similar strategy using Cas9 has been demonstrated to increase rice resistance to Xanthomonas oryzae by disrupting the locus where pathogen effectors bind, thereby reducing pathogen virulence (Jiang et al., 2013).
Selectable marker genes (SMG) are used to select genetic transformants, preferentially during transgenesis. SMGs encode proteins to help transformants detoxify antibiotics or herbicides during selection. However, presence of SMG in transgenic foods and feed products is a major concern in GMO production. Regulatory agencies and consumers are concerned regarding SMG that may negatively impact the environment and affect the health of humans and animals. It is desirable to eliminate the SMGs after their use. SSNs such as ZFNs have been used in gene deletion (Yau and Stewart 2013), and a similar approach can be used for SMG deletion using CRISPR-Cas9.
Although the use of HDR-mediated genome editing through CRISPR-Cas9 to improve crop quality is still at its infancy, the technology is especially promising in the area of developing herbicide tolerance in crops. The enzyme involved in biosynthesis of aromatic amino acids, 5-enolpyruvylshikimate-3-phosphate synthase (EPSPS), is inhibited by the herbicide, glyphosate. Similarly, acetolactate synthase (ALS) involved in branched chain amino acid biosynthesis is also a target for several herbicides (imidazolinones and sulfonylureas). Modifying the domains in the plant enzymes EPSPS and ALS, which interact with corresponding herbicides, would confer herbicide tolerance in the plant (Voytas and Gao, 2014). A proof of concept study with ZFN has successfully demonstrated modification in the ALS gene resulting in plant herbicide tolerance (Townsend et al., 2009). Computational methods predict precise amino acid substitution within the glutathione S-transferase (GST) would direct its activity against a diverse set of herbicides (Govindarajan et al., 2015). Although bacterial transgenes have been used in commercially available herbicide tolerant crops, the use of genome engineering would provide the distinct advantage of circumventing the use of transgene. Transiently expressed Cas9 system can be employed to generate genome engineered crops having such precise amino acid substitutions.
Orthologous multiplex genome engineering platform of the CRISPR system is a boon for plant biologists studying metabolic pathways, transport, and polygenic traits. It has been suggested that the multiplex genome editing approach holds promise in efforts to increase understanding of the mechanisms of auxin transport in plants (Balzan et al., 2014). Chemically-induced mutations in the florigen-associated plant flowering pathway offers the possibility of customizing plant architecture that translates to enhanced yield, as can be seen in the study demonstrating improvement in productivity of tomato (Park et al., 2014). Another possibility of the use of SSN is in the directed mutagenesis of the genes in the florigen pathway pinpointing an optimal balance of florigen phytohormone. This has immense promise in terms of crop yield enhancement.
Most genotypes of rice exhibit complete resistance to rust, directing non-host resistance to rust pathogens (Ayliffe et al., 2011). Identification and transfer of these resistance (R) genes to other cereal crops is a very promising area of research. Analyzing and mining data concerned with seed genomics is considered a herculean task for plant biologists. Multiplex genome engineering is predicted to play an important role in the future for dissecting the various regulatory networks involved in seed development (Becker et al., 2014). Similar approaches can be directed at elucidating the diverse plant signaling and response cascades involved with abiotic and biotic stresses, which can be modulated by genome engineering for stress management, toughening up crop plants against a wide array of stresses.
In conventional plant transformation, each transgenic event results from a random and unpredictable insertion of a transgene into the genome. As a result, each event is predisposed to independent evaluation of performance. HDR mediated by Cas9 gives transgenic technology a new edge by providing the ability to target insertion of transgene to desired locations within the genome. This breakthrough capacity results in uniform transgenesis events by directing transgenes to ‘safe spots' that avoid integration into essential loci which can disrupt important gene functions. These suitable regions in genomes, denoted as safe harbor loci, have already been investigated and identified in rice (Cantos et al., 2014).
In 1995, the bel (bentazon sensitive lethal) mutation in rice was first created through radiation mutagenesis (350 Rad Co60-γ-ray). The bentazon-lethal rice mutants are susceptible to bentazon and sulfonylureas herbicides. These mutants are used for production of hybrid rice, to prevent contamination of selfed seeds within hybrid seed lot. Spraying specific herbicide during the seedling stage will eliminate progeny from selfed plants. Xu et al. used CRISPR-Cas9 technology to knockout the BEL gene through Agrobacterium- mediated transformation in rice with mutagenesis efficiency between 2% and 16%. The bi-allelic BEL knock-out lines showed sensitivity to bentazon (Xu et al., 2014).
SSNs can also be an essential tool for biofortification. Trait development for nutritional enhancement follows two approaches: (1) Enhancing the amount of nutrients in the foodstuff, and (2) eliminating the anti-nutrients that reduces the food quality or nutrient bioavailability. In plants, major essential amino acids such as lysine, methionine, and tryptophan have their own biosynthetic pathways, which are regulated by feedback inhibited enzymes. Transgenic crops with increased essential amino acid content are generated by incorporating bacterial biosynthetic enzymes that are free of feedback inhibition (Ufaz and Galili, 2008). GM crops developed by this method must have to undergo the extensive regulations, which reduce their commercial viability. Genome editing holds promise in this regard for metabolic engineering of essential amino acid either by (1) modifying domains responsible for regulating feedback inhibition, or by (2) knocking out enzymes involved in catabolism of specific amino acids, thereby increasing amino acid content (Ufaz and Galili, 2008).
Eliminating anti-nutrients enhances bioavailability of nutrients for absorption into our system. Phytic acid is a key anti-nutrient that forms strong complexes with minerals such as iron, making it unavailable for absorption. Biosynthetic enzymes involved in phytic acid synthesis have been targeted with various approaches for increasing nutritional quality. Maize lines knocked out for the gene 1,3,4,5,6-pentakisphosphate 2-kinase (IPKI) coding for an enzyme involved with the final step of phytate biosynthesis, has been generated through ZFN mutagenesis (Shukla et al., 2009). A similar approach could be followed for increasing the bioavailable calcium in plants by the reduction of oxalate biosynthesis. Cyanogenic glucosides, linamarin and lotaustralin in tapioca can be reduced by suppression of their biosynthetic genes (Jørgensen et al., 2005). Similarly, gliadins could be knocked-out with CRISPR-Cas9 system, as a strategy for developing gluten-free diet for celiac disease patients (Gil-Humanes et al., 2014).
Cas9-based genome editing has already been successfully employed in goat (Ni et al., 2014), sheep (Fan et al., 2014), pig (Tan et al., 2013), and cattle (Tan et al., 2013) (Table 2). There is promise for improving livestock quality in the future by the use of this technology. Biofortification of milk through the knockout of allergenic gene such as β-lactoglobulin in genome-engineered dairy animals might be possible in the future. Although distant, potential of this technology for improving the quality of agricultural produce as well as livestock remains massive.
Use of Cas9 sequence specific nucleases is especially exciting in the field of agriculture because these targeted modifications in crops from transient delivery of SSN are indistinguishable from naturally occurring mutants or those generated from chemical and physical mutagenesis (Voytas and Gao, 2014). Transient delivery of SSNs through agroinfiltration, viral vectors, or physical methods such as biolistics, may also be employed. NHEJ-induced knockouts of genes are indistinguishable from chemical or physical mutagenesis, resulting in nontransgenic-like plants containing stably inherited desired sequence changes (Jiang et al., 2014). This potential will necessitate a different regulatory framework for covering genome engineered crops. The USDA has discreetly stated that genome modified crops with ZFN fall outside their regulatory authority. Dow Agrosciences were assured by the USDA of not requiring regulatory oversight for the corn developed through ZFN based genome editing (Waltz, 2012). However, HDR-mediated modification through engineered nucleases in plants is considered as GMOs because the nucleic acids are delivered and incorporated into the genome. The HDR modifications would come under the same category as GMOs for biosafety considerations in countries that adhere to process-based biosafety regulation.
CRISPR-Cas9: Biomedical Advancements and Opportunities
Genome editing enables the rapid generation of cellular and animal models. Disease mutations are studied by using models or by making a phenocopy of a particular disorder (Sander and Joung, 2014). Genome-wide association studies (GWAS) have found several regions in the genome that harbor potential risks for polygenic diseases such as diabetes, Alzheimer's, schizophrenia, and autism. Cas9 based multiplex genome engineering holds promise in assessing the roles of these loci, both individually and simultaneously. Effects of genome modifications can be tracked by genome editing of stem cells, followed by their differentiation into cell type of interest (Hsu et al., 2014). Cas9 mouse lines to express Cas9 in a constitutive or tissue specific manner have been generated by crossing a Cre-depended Cas9 mouse with specific Cre-driver strains. Delivery of specific gRNA to the Cas9 mice enabled both ex vivo and in vivo genome editing of neurons, immune cells, and endothelial cells. Simultaneous modeling of lung adenocarcinoma through multiplexing has also been demonstrated (Platt et al., 2014).
Genome engineering holds great promise for regenerative medicine-based therapeutics. Direct genome editing in tissues can be a primary route for treatment. Several proof of concept studies have proposed such methods for correcting monogenic recessive genetic disorders such as hemophilia (Li et al., 2011), cystic fibrosis (Schwank et al., 2013), Duchenne muscular dystrophy (Ousterout et al., 2013), tyrosinemia (Yin et al., 2014), Fanconi anemia (Osborn et al., 2014), and sickle cell anemia (Sun and Zhao, 2014). Inactivation of a mutant allele via genome editing has been proposed for correcting dominant negative genetic disorders such as retinitis pigmentosum and transthyretin-related hereditary amyloidosis (Hsu et al., 2014). Variations in the non-genic (enhancer) regions have been shown to underlie autoimmune diseases, making DNA manipulations through genome editing a promising therapeutic intervention for their mitigation (Farh et al., 2014).
Besides repairing disorder associated genes, genome editing-based regenerative medicine can also be used to protect individuals from disease risk by disrupting certain genes. Proof of concept studies in mice has shown that disrupting the Pcsk9 gene (PCSK9 is an antagonist to the LDL receptor) in vivo (liver) has therapeutic promise against cardiovascular disease in humans (Ding et al., 2014). Successful clinical trials against HIV infection have been reported through the use of SSNs (ZFN) for disruption of the CCR5 encoding gene required by HIV-1 for entry into host. Viral infection is crippled because of disrupted receptor activity in the CD4+ cells. These results were derived from genome edited stem cells that were transplanted into patients. Similar results have been obtained in HIV-inoculated cell cultures as well (Tebas et al., 2014; Ye et al., 2014). Engrafting Cas9 modified CCR5− human hematopoietic stem and progenitor cells is a promising approach in combating AIDS (Mandal et al., 2014).
Targeted homing devices for manipulation of transcriptional networks and epigenetic landscapes is more favorable for regulating cell fate than naturally occurring activators, since targeted homing devices are free from endogenous regulatory and signaling elements (Eguchi et al., 2014). Current strategies employ generating iPS from patient-derived adult cells and differentiating them into cell type of interest using regulatory elements like miRNA and TFs (Chanda et al., 2014; Victor et al., 2014). The advent of targeted genome engineering allows DNA homing devices fused with regulators to perform tasks with an ease and efficiency which can improve regenerative medicine by leaps and bounds.
The Cas9 system has also been demonstrated to have potential in antimicrobial therapies that re-sensitize bacteria to antibiotics. This can be used to modulate virulence of bacterial populations. Phages and conjugative plasmids enabled delivery of SSNs to microbial populations, using Cas9 programmed to target specific sequences underlying antibiotic resistance and virulence in bacteria (Citorik et al., 2014). A study has revealed the possibility of engineering heritable resistance in cattle towards Mycobacterium bovis (tuberculosis) by HDR-mediated knock-in of mouse gene SP110 using TALEN nickases (Wu et al., 2015). Malarial parasite Plasmodium falciparum has been notoriously resistant to efforts of the research community to elucidate its intra-erythrocytic developmental genetics, slowing down the development of novel drugs and vaccines. CRISPR-Cas9 has emerged as a fast and efficient tool that has been successfully applied in this regard to manipulate or knockout malaria genes (Ghorbal et al., 2014; Wagner et al., 2014), which had taken extensive time previously.
Conclusion
Signifying the importance of translational research, CRISPR-Cas9 technology has witnessed a greatly accelerated development from the role in bacterial immunity to therapeutics and crop improvement. Its speedy adoption has been made possible by the online user platforms and open source distribution. Prior to clinical translation of Cas9, its safety and physiological effects still need to be thoroughly assessed and characterized. Concerned authorities must determine whether CRISPR-Cas9 based genome engineering technology applied to crops merits an overall exemption from the regulatory process or a less rigorous regulation.
The ability of this review to bring such a vast and diverse area under one umbrella indicates the unified nature of biology at the molecular level, which is exactly how CRISPR system functions. It integrates the target which mostly is DNA, the RNA that directs the activity (gRNA), and the protein Cas9 enzyme. The CRISPR-Cas system has this unique ability to bring together all the three molecules of life in a customizable manner.
Footnotes
Acknowledgment
We thank Mona Easterling for her critical proofreading of the manuscript.
Author Disclosure Statement
The authors declare that they have no competing interests.