
Abstract
Abstract
Increasing drug resistance in Plasmodium falciparum is an important global health burden because it reverses the malarial control achieved so far. Hence, understanding the molecular mechanisms of drug resistance is the epicenter of the development agenda for novel diagnostic and therapeutic (drugs/vaccines) targets for malaria. In this study, we report global comparative transcriptome profiling (RNA-Seq) to characterize the difference in the transcriptome between 48-h intraerythrocytic stage of chloroquine-sensitive and chloroquine-resistant P. falciparum (3D7 and Dd2) strains. The two P. falciparum 3D7 and Dd2 strains have distant geographical origin, the Netherlands and Indochina, respectively. The strains were cultured by an in vitro method and harvested at the 48-h intraerythrocytic stage having 5% parasitemia. The whole transcriptome sequencing was performed using Illumina HiSeq 2500 platform with paired-end reads. The reads were aligned with the reference P. falciparum genome. The alignment percentages for 3D7, Dd2, and Dd2 w/CQ strains were 85.40%, 89.13%, and 84%, respectively. Nearly 40% of the transcripts had known gene function, whereas the remaining genes (about 60%) had unknown function. The genes involved in immune evasion showed a significant difference between the strains. The differential gene expression between the sensitive and resistant strains was measured using the cuffdiff program with the p-value cutoff ≤0.05. Collectively, this study identified differentially expressed genes between 3D7 and Dd2 strains, where we found 89 genes to be upregulated and 227 to be downregulated. On the contrary, for 3D7 and Dd2 w/CQ strains, 45 genes were upregulated and 409 were downregulated. These differentially regulated genes code, by and large, for surface antigens involved in invasion, pathogenesis, and host–parasite interactions, among others. The exhibition of transcriptional differences between these strains of P. falciparum contributes to our understanding of the attendant, drug-sensitivity phenotypes, and by extension, the current efforts in maintaining global health by developing novel diagnostics and therapeutics for malaria.
Introduction
M
The antimalarial drugs are classified into three broad groups (quinolone, antifolate, and artemisinin derivatives) based on its mode of action (Antony and Parija, 2016). The quinolone derivatives bind to the β-hematin molecule that is a by-product of hemoglobin degradation and inhibit the heme detoxification pathway in the digestive vacuole of the parasite. Chloroquine, amodiaquine, mefloquine, quinine, and so on are some of the few examples of quinolone compounds. The antifolate derivatives bind to the Phdhps and Pfdhfr genes that are involved in the folate biosynthesis pathway in the cytoplasm of the parasite. The antifolate compounds include pyrimethamine and sulfadoxine. The artemisinin derivates belong to the endoperoxide family and include artesunate, artemisinin, and artemether. The mechanism of action of artemisinin drug remains unclear. However, it is believed that artemisinin acts by releasing free radicals from the endoperoxide bridge during the heme-mediated decomposition (Meshnick, 2002) These free radicals damage the susceptible proteins and thereby kill the malaria parasites.
The emergence of resistant parasites against known antimalarial drugs has diminished the global eradication efforts, and hence, the reemergence of malaria as a major health issue worldwide (Arav-Boger and Shapiro, 2005). The mechanism of drug resistance in Plasmodium is not fully understood. However, genetic markers have been identified to detect the drug resistance based on the polymorphisms in the genes involved such as the P. falciparum chloroquine resistance (CQR) transporter, PfCRT (chloroquine) (Fidock et al., 2000); dihydropteroate synthase, PfDHPS (sulfa drugs) (Gregson and Plowe, 2005); dihydrofolate reductase, PfDHFR (pyrimethamine) (Peterson et al., 1988); cytochrome b (atovaquone) (Korsinczky et al., 2000); and kelch 13 propeller protein (artemisinin) (Ariey et al., 2014). Although the polymorphism in these genes contributes to drug resistance, there is a lack of sufficient information to explain resistance completely as its effects vary with concentration of drug and strain genotype (Sidhu et al., 2002).
Hence, to overcome the selective antimalarial drug pressure, different strains of P. falciparum may also depend on different mechanisms for regulation of genes, such as overexpression/repression of genes against drug pressure and stimulation of different regulation pathways to evade the compound effects. For example, increased copy number of P. falciparum multidrug resistance gene, PfMDR1, is associated with mefloquine resistance that increases the cellular transcripts produced (Price et al., 2004). Thus, gene regulation of P. falciparum, in response to drug exposure, may either function independently or in association with previously identified polymorphisms that support the endurance of the parasite survival.
P. falciparum has a 22.8 Mbp sized, high AT-rich (∼80%) genome, comprising 14 linear chromosomes, a circular plastid, and a linear mitochondrial genome. The whole-genome sequence (WGS) of P. falciparum 3D7 strain was sequenced as part of the multicentric consortia representing the Sanger Centre, The Institute for Genomic Research (TIGR), and Stanford University. This WGS analysis resulted in the identification of ∼5400 genes present in the genome, of which more than 60% of the predicted genes encoding for proteins lack sequence similarity or orthologs in other different organisms (Gardner et al., 2002). Whole-genome gene expression profiling methods help to explore gene functions and new possibilities in malaria research (Bozdech et al., 2003b; Llinas and DeRisi, 2004). Gene expression studies of P. falciparum throughout the intraerythrocytic development stage, using DNA microarray (Bozdech et al., 2003a, 2003b; Llinás et al., 2006) and proteomic approach (Foth et al., 2008, 2011), demonstrated that a continuous cascade of genes was expressed to regulate the progression of the parasite from invasion to replication in the erythrocytes.
Previous studies have attempted to sequence P. falciparum RNA transcripts based on expressed sequence tags (Lu et al., 2007) and have investigated a small fragment of cDNA sequences from Plasmodium spp. (Wakaguri et al., 2009a, 2009b), resulting in low-resolution data due to lack of information in gene model predictions of P. falciparum. However, these transcriptome data in P. falciparum suggested the prevalence of versatile length of untranslated regions (UTRs) and shift in the splicing pattern of the genes. In 2008, high-throughput sequencing techniques using RNA-Seq were reported to sequence the expressed RNA transcripts in human, mouse, and yeast (Mortazavi et al., 2008; Nagalakshmi et al., 2008; Pan et al., 2008; Sultan et al., 2008; Wilhelm et al., 2008). Annotation of genes, analyzation of UTR regions, validation of new and preexisting splice patterns, and detection of new transcripts can be done reliably using the RNA-Seq approach (Wang et al., 2009). In 2010, RNA-Seq was performed for the intraerythrocytic development cycle of P. falciparum to understand the association of the RNA transcripts expressed and to determine the splicing dynamics that happen during the growth of the parasite (Otto et al., 2010). Although several profiling methods have been used to study the biology of P. falciparum, there are insufficient data to determine the mechanism of drug resistance.
In this study, we have performed the whole transcriptome sequencing at the 48-h intraerythrocytic stage between laboratory-adapted P. falciparum 3D7 and Dd2 strains. Chloroquine effectively kills the parasite at the trophozoite stage than at a schizont and merozoite stage (Slater, 1993), which is predominantly present at 48 h of the intraerythrocytic stage. These strains have distinct drug sensitivity phenotypes, where 3D7 strain is sensitive to chloroquine and resistant to sulfadoxine, while the Dd2 strain is resistant to both chloroquine and mefloquine (Rathod et al., 1997), and these strains have originated from distant geographical regions, the Netherlands for 3D7 strain and Indochina for Dd2 strain. This study aimed to investigate the strain-specific phenotypes and the mechanism of gene regulation involved in chloroquine drug resistance using the whole transcriptome sequence profiling.
Materials and Methods
Parasite cell culture
P. falciparum 3D7 (chloroquine-sensitive [CQS]) and Dd2 (CQR) strains were gifted by Dr. Paushali Mukherjee, Malaria Laboratory, International Centre for Genetic Engineering and Biotechnology (ICGEB), New Delhi. These strains were maintained by continuous in vitro culture as described previously by Trager and Jensen (1976). Briefly, these strains were maintained in “O” positive red blood cells at 1–5% parasitemia and 5% hematocrit in RPMI 1640 media (Gibco) supplemented with 5% AlbuMAX (Gibco), 0.05% hypoxanthine (Sigma), 25 mM HEPES buffer, 2.1 g/L sodium bicarbonate, and 200 μg/mL gentamycin. Parasite cells were synchronized using 5%
RNA extraction, library construction, and sequencing
Total RNA was extracted from the parasitized cell pellets using the RNAiso Plus reagent (Takara, Japan), following the manufacturer's protocol. The concentration and quality of the extracted RNA were measured using NanoDrop, Qubit, and 2100 Bioanalyzer (Agilent Technologies). The RIN values for 3D7, Dd2, and Dd2 w/CQ strains were 8.4, 8.5, and 8.2, respectively, confirming that the quality of RNA was good for cDNA library preparation. cDNA libraries were constructed following the TruSeq RNA Sample Preparation Kit v2 low sample protocol (Illumina, Inc.), according to the manufacturer's guidelines. The cDNA library quality was measured using the 2100 Bioanalyzer, and whole transcriptome sequencing was performed using HiSeq 2500 platform (Illumina, Inc.) with paired-end reads and 2 × coverage (Kukurba and Montgomery, 2015).
Transcript assembly and annotation
The quality of the fastq files was accessed using the FastQC tool (Babraham Bioinformatics), which represents the data quality based on base quality score, sequence quality score distribution, GC distribution, and so forth. The reads having a Phred quality score of more than 30 were used for further analysis. The RNA contaminations (i.e., rRNA, ncRNAs) were also removed using Bowtie2 (version 2.1.0), in-house Perl scripts, and Picard tools (version 1.85). The processed reads were then aligned to the reference P. falciparum genome (from Ensemble database Release 26) using TopHat program (version 2.0.8) with default parameter (Kukurba and Montgomery, 2015; Trapnell et al., 2013). The paired-end reads generated for the samples were submitted with the accession no PRJNA308455 in the BioProject database of NCBI (Antony et al., 2016).
Transcript expression and differential expression analysis
The aligned reads were used to estimate the expression of the genes and transcripts using cufflinks program (version 2.0.2) with default parameters (Trapnell et al., 2013). The FPKM value ≥0 was considered as a cutoff for the expression of the transcripts present in the sample. Cuffdiff program has been used to determine the differentially expressed genes between the sensitive and resistant strains with the p-value ≤0.05. The differential expression of genes was validated by comparing with previous transcriptome data analysis of P. falciparum 3D7 and Dd2 strains (Bozdech et al., 2003a, 2003b; Llinás et al., 2006). The RNA-Seq expression study data were deposited in Gene Expression Omnibus functional genomics data repository of NCBI with the accession no GSE77499 (Antony et al., 2016). The up- and downregulated genes were further characterized and enriched based on the KEGG pathway and gene ontology (GO) enrichment for cellular component, biological, and molecular function using PlasmoDB database (Aurrecoechea et al., 2009).
Results
RNA-Seq data analysis
To understand the interstrain variability of CQS and resistant strains of P. falciparum, we characterized the transcriptome for the laboratory-adapted 3D7 and Dd2 strains at the 48-h intraerythrocytic stage with 90% trophozoites. Chloroquine kills the trophozoite stage of parasites more effectively than the schizont and merozoite stage. Total RNA extracted from the harvested parasite cells was converted to cDNA after the isolation of mRNA and was purified. The cDNA library generated was sequenced using Illumina HiSeq 2500 platform with 100 × 2 paired-end reads. It produced 21,942,058, 20,001,304, and 14,745,529 paired-raw reads for 3D7, Dd2, and Dd2 w/CQ strains, respectively, generating nearly 4.3, 4, and 3 Gb raw data (Table 1). The fastq file (raw reads) of the samples showed ∼93% of total read passed Phred score with Q ≥ 30. The distribution of low GC content (∼30%) for the three samples is due to the AT-rich genome sequence of P. falciparum (Gardner et al., 2002).
GC.
The base composition and base quality plots were used for checking the quality of the sample sequence fastq file and to verify whether there is any bias in the read. The base composition exhibited a slight bias at the start of the reads and also a slight drop in base quality was observed at the start of the reads. The low-quality bases were trimmed from the raw reads to retain only the high-quality reads for sequence analysis. The preprocessed reads were aligned to the reference P. falciparum gene model using the TopHat program based on pair-wise alignment. The TopHat program produced data with an alignment percentage of 85.40%, 89.13%, and 84% for 3D7, Dd2, and Dd2 w/CQ strains, respectively.
After alignment with reference, P. falciparum genome, cufflinks program was used to estimate the expression of the genes and transcripts. The comparative gene expression distribution plot for the three P. falciparum samples is shown in Figure 1. For quality control measurement, dispersion estimation plot is drawn to estimate overdispersion for each sample and principal component analysis plot to check the clustering and explore the relationships between the conditions. A comparative study of all the three P. falciparum strains found 4959 common protein-coding genes across the samples and also estimated 52, 10, and 8 unique protein-coding genes expressed in samples 3D7, Dd2, and Dd2 w/CQ, respectively (Fig. 2). The functional annotation of the expressed genes is done using the UniProt database information. The functions of the gene expressed were determined based on the gtf file information that is available for the species, as depicted in Figure 3A. The gene expression estimation for the samples is provided in Supplementary Tables S1–S3.
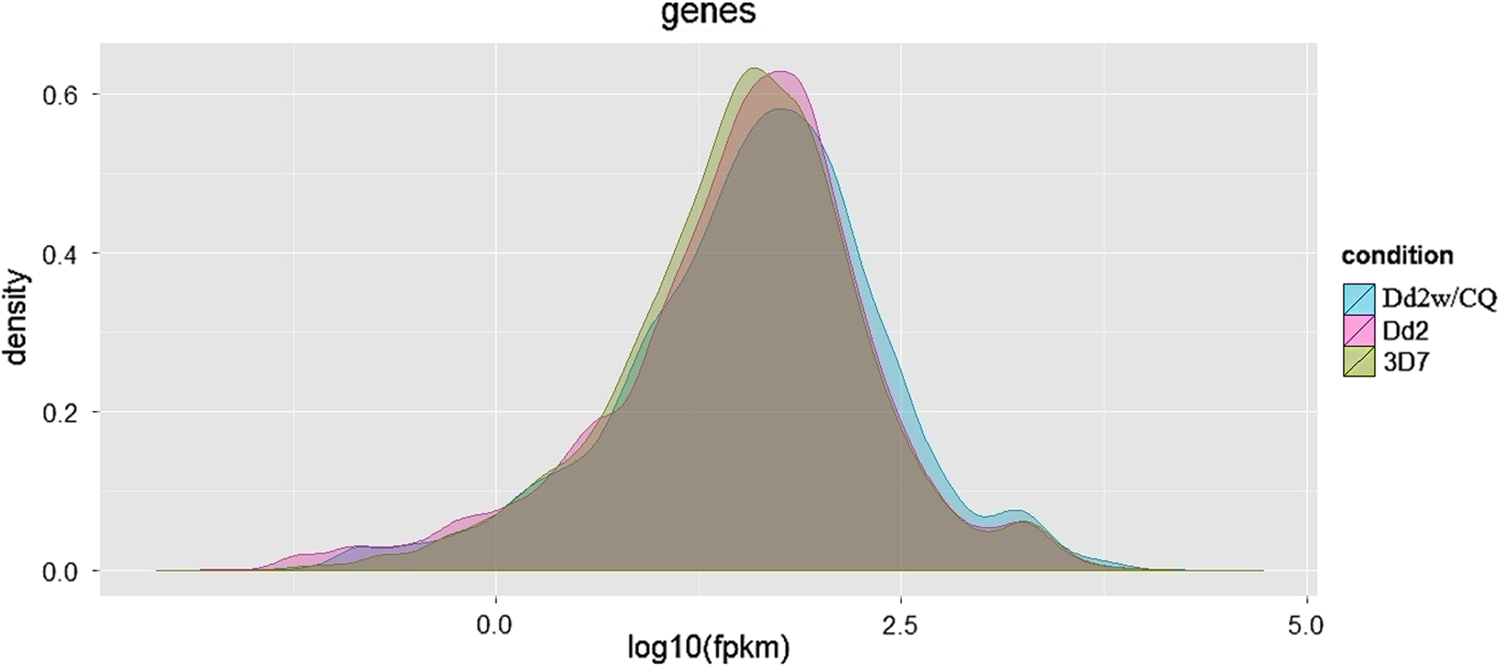
Comparative gene expression distribution plot between the three Plasmodium falciparum samples.
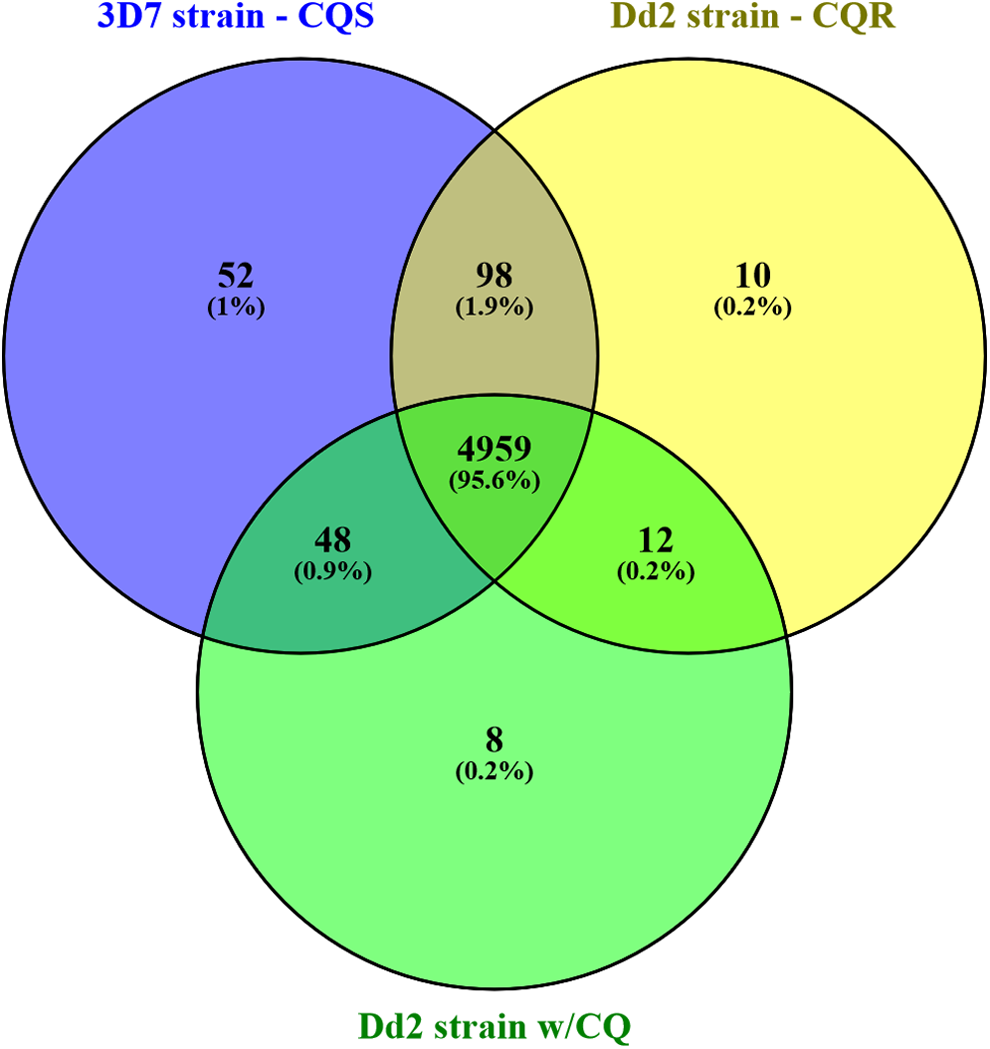
Venn diagram of the protein-coding genes in the 3D7 (chloroquine sensitive), Dd2 (chloroquine resistant), and Dd2 w/CQ (cultured in the presence of CQ) strains. CQ, chloroquine.

More than 60% of the genes are hypothetically proteins with unknown function and are uncharacterized. Thus, ∼40% of the genes are protein coding with known functions and are characterized. Approximately 5% of the genes were found to act as transporter and were involved in the transmembrane transport of metabolite, drugs, and so on. Also, ∼5% of genes are involved in protein translation mechanism, and ∼4% of genes are associated with RNA metabolism/transcription process. Lack of expression in genes, such as knob-associated histidine-rich protein (KAHRP, PFB0100c) and erythrocyte membrane protein 3 (PfEMP3, PFB0095c), was detected in Dd2 and Dd2 w/CQ strains when compared to 3D7 strain. These genes are unique to Dd2 strain and could be due to the highly polymorphic regions in its genome (Llinás et al., 2006).
Surface antigen expression profiles
Moreover, ∼3% of protein-coding genes are surface antigens and are involved in antigenic variation, pathogenesis, and host–parasite interaction. The surface antigens are classified into three major families, namely, the var genes (erythrocyte membrane proteins, PfEMP1), the rifins gene family (repetitive interspersed proteins), and the stevors genes (subtelomeric variant open reading frames). Differences in the gene expression were observed primarily in the surface antigens between the CQS and resistant strains of P. falciparum (Fig. 3B). P. falciparum regulates a vast repertoire of surface antigens and evades the host immune response during the parasite's successive generations (Kyes et al., 2001). Also, in a previous transcriptome study by DNA microarray, a different cohort of var genes was expressed completely and demonstrated that even laboratory-adapted strains utilize a different set of surface antigen-related genes in their developmental cycle (Le Roch et al., 2003; Llinás et al., 2006). Thus, the difference in the expression of surface antigens in P. falciparum varies even in in vitro culturing.
Differentially expressed genes in P. falciparum strains
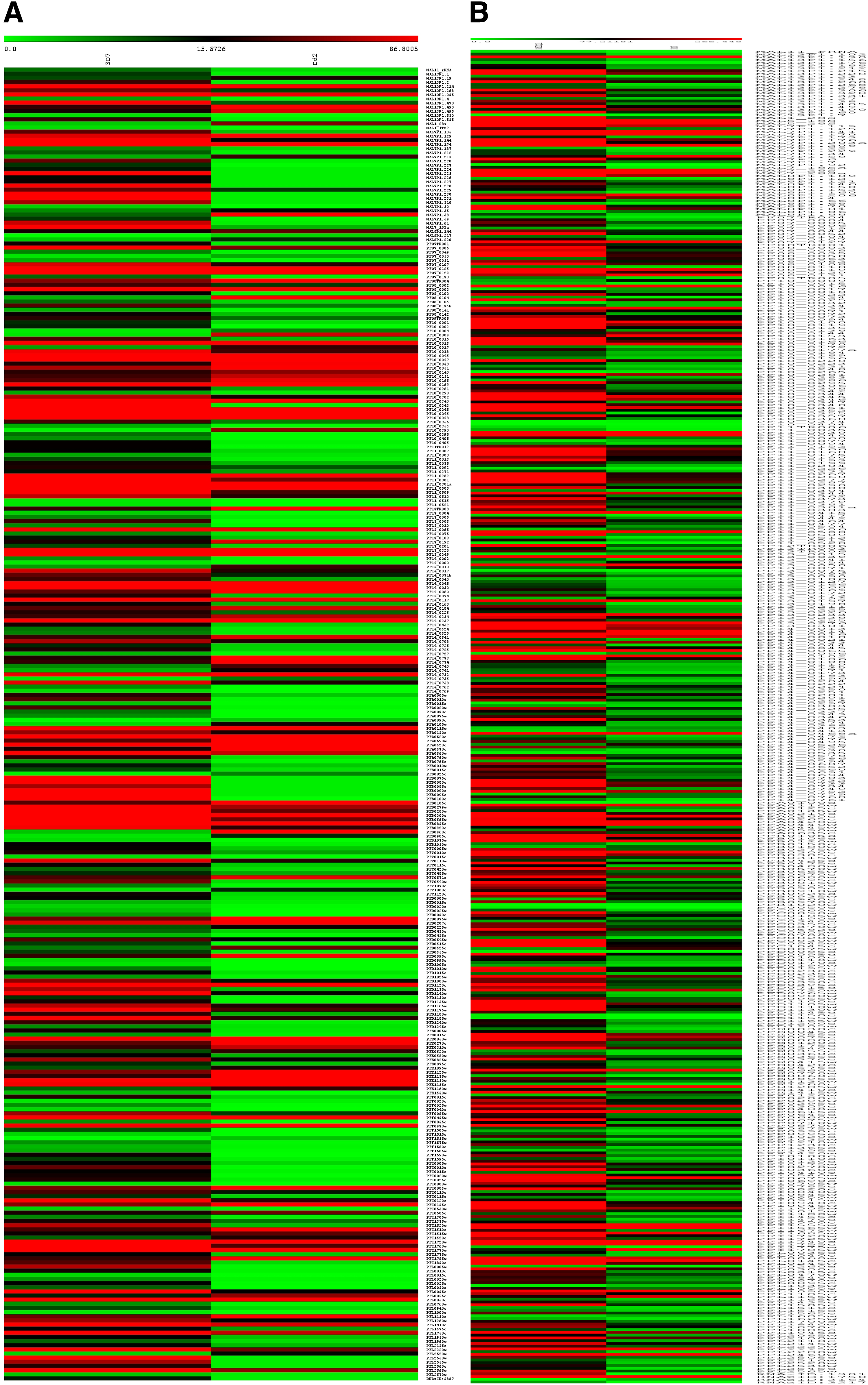
The correlation scatter plot and distance matrix plot were estimated to check the pair-wise similarities among the three samples of P. falciparum. The differential gene expression analysis between the CQS (3D7) and resistant (Dd2 and Dd2 w/CQ) strains was performed using cuffdiff program of cufflink package with default settings. The differentially expressed genes with the p-value cutoff ≤0.05 have detected 89 upregulated and 227 downregulated genes between 3D7 and Dd2 strains, while 45 upregulated and 409 downregulated genes between 3D7 and Dd2 w/CQ strains. Figure 4 depicts the differentially expressed genes, based on chromosome-wise coverage of Dd2 and Dd2 w/CQ strains with reference to 3D7 strain. The highly differential regulated genes were detected in chromosomes 10–14 of Dd2 w/CQ strain, with the highest in chromosome 14. Heat map of all the differential expression comparisons for the p-value cutoff ≤0.05 is shown in Figure 5. Most of the differentially regulated genes were involved in invasion, cell–cell adhesion, pathogenesis, surface antigens, and so forth. This result is in agreement with the previous findings that gene expression changes among 3D7, Dd2, and HB3 strains were primarily detected in the loci of the encoding surface antigens involved in immune evasion (Llinás et al., 2006).
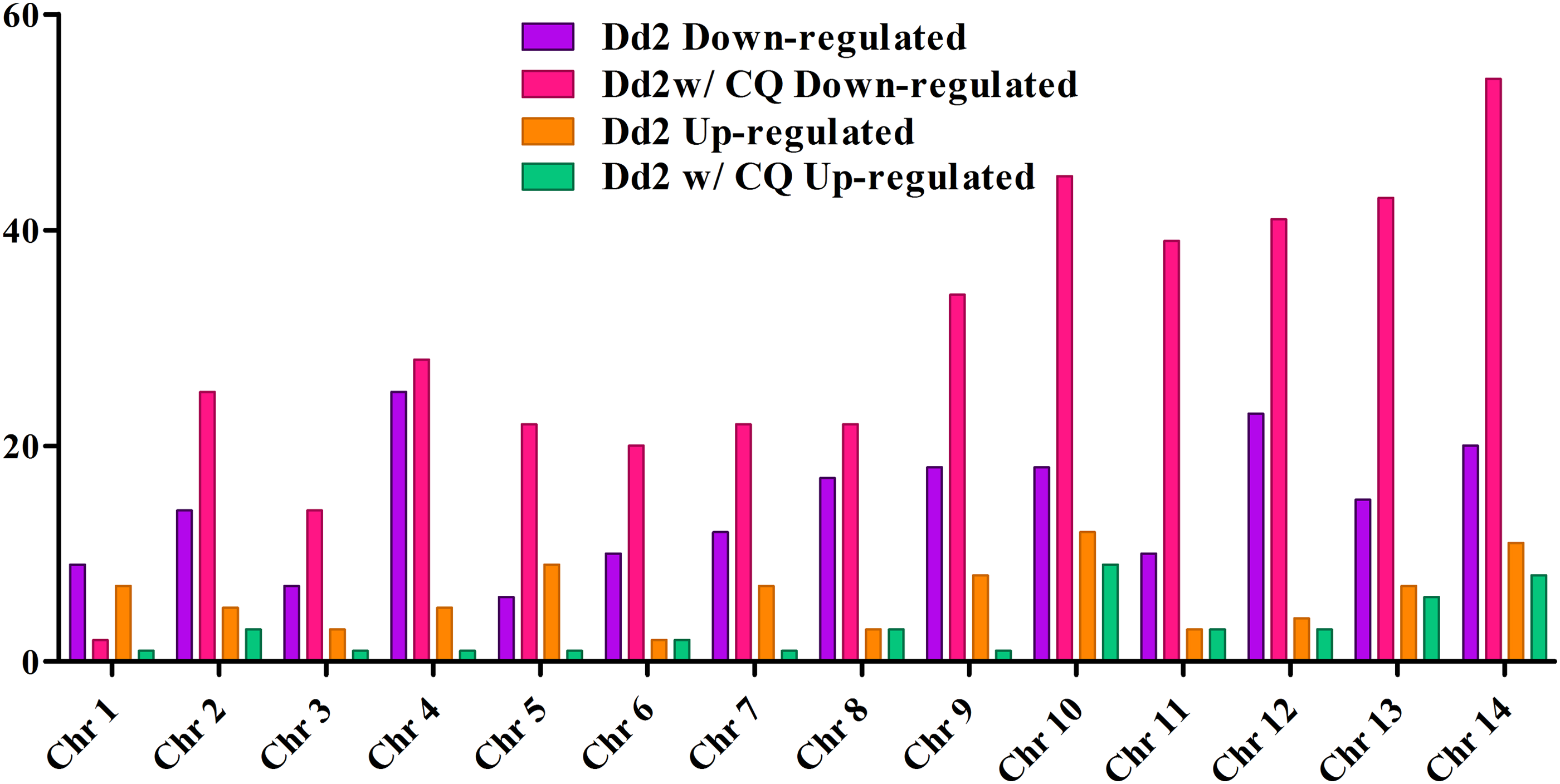
Distribution of differentially expressed protein-coding genes across the P. falciparum genome.
The differentially expressed genes were further characterized based on GO enrichment and KEGG pathway using PlasmoDB (Supplementary Table S4–S7). The GO enrichment of the downregulated genes of Dd2, based on molecular function, with a p-value cutoff 0.05, exhibited functions involved in cell adhesion binding, host cell surface binding, receptor binding, protein binding, and protein kinase activity mainly. In addition to these molecular functions, the downregulated genes of Dd2 w/CQ strain exhibited carbohydrate and heparin binding, lipid metabolism, and lipase and phosphatase activity. Similarly, the GO enrichment of the upregulated genes of Dd2 strain described the molecular functions involved in amino acid binding, DNA binding, and DNA metabolism, with a p-value cutoff 0.05. The upregulated genes of Dd2 w/CQ exhibited transferase activity and hydrolase activity, in addition to the functions described for Dd2 upregulated genes. Also, based on KEGG pathway analysis using PlasmoDB with a p-value cutoff 0.05, these downregulated genes are involved in lipid metabolism and nucleotide and amino acid metabolism, whereas the upregulated genes are involved in nucleotide and lipid metabolism.
Discussion
RNA-sequencing is an invaluable tool in the field of genome sequencing project, as it helps to identify genes, gene annotation, splicing pattern, and so on. Also, it helps in the comparison of gene expression analysis between the reference and test model (Kukurba and Montgomery, 2015). The most virulent parasite, P. falciparum, has evolved in its virulence factors and drug resistance; this is responsible for a wide range of interstrain-specific differences worldwide. However, strain-specific phenotypic differences are poorly characterized in P. falciparum, with an exception in the detection of the molecular marker for drug resistance. This study helps to understand the gene expression pattern of the commonly used 3D7 and Dd2 strains and to identify regulatory pathways by comparing the 48-h trophozoite stage transcriptome using RNA-Seq. The main differences between these two strains are their distant geographical regions and distinct drug sensitivity.
In this study, transcriptome analysis was carried out to determine the differential gene expression between the laboratory-adapted, CQS, and resistant strains of P. falciparum 3D7 and Dd2, respectively. Also, P. falciparum Dd2 strain grown in the presence of MIC 160 nM CQ was included in the transcriptome sequencing to study its gene expression profiling. The parasite degrades the hemoglobin molecule in the food vacuole of the trophozoite for its growth and survival. Chloroquine kills the parasite by preventing hemoglobin degradation into hemozoin pigments, which is harmful to the parasite. Trophozoite stage (late ring and mature trophozoite stage) of the parasite was highly sensitive to the chloroquine when compared to the mature schizonts and merozoites (Slater, 1993). Thus, the susceptibility of the chloroquine drug is in accordance with the hemoglobin catabolism in the parasite. In this study, the parasite cells were grown by the in vitro culture method and harvested at the 48-h intraerythrocytic cycle with ∼90% trophozoites in the erythrocytes.
This study indicates that the overall gene expression pattern of the 48-h transcriptome profiling of P. falciparum strains 3D7, Dd2, and Dd2 w/CQ is notably the same for a majority of the genes. This preservation of gene expression describes that most of the key processes are well conserved and maintained among strains (Llinás et al., 2006). Also, the absence of KAHRP and PfEMP3 gene expression in Dd2 strain indicates that the genes are conserved and this is explained by the presence of highly polymorphic loci of Dd2 strain genome (Llinás et al., 2006). Major changes in gene expression were observed for surface antigens involved in pathogenesis and immune evasion. These changes in surface antigen expression could be because of strain-specific mutational effects, such as polymorphisms, deletions, or gene silencing, as suggested by Llinás et al. (2006). The regulation of var gene in telomeric regions has been demonstrated by Sir2-dependent silencing (Duraisingh et al., 2005; Freitas-Junior et al., 2005), and our result also indicates that similar gene silencing might have occurred in other regions of the genome in a strain-specific manner.
We expected a differential expression, between the sensitive and resistant strains, of the genes involved in drug resistance. CQR is mainly attributed to the point mutation in the Pfcrt gene that prevents the chloroquine accumulation in the food vacuole of the parasite (Fidock et al., 2000). Also, polymorphisms in the Pfmdr1 and Pfmrp genes have shown to be involved in CQR in association with Pfcrt gene (Mu et al., 2003; Reed et al., 2000). We detected no significant changes in the up- or downregulation of these genes involved in drug resistance between the 3D7 and Dd2 strains. Previous findings suggest that cells grown under favorable conditions might not be under environmental stress, which tends to alter the gene expression (Llinás et al., 2006; Myrick et al., 2003). Hence, the Dd2 strain was cultured in a condition containing chloroquine to find changes in expression of these genes. We detected differences in expression of genes involved in resistance genes between the sensitive and resistant strains, but are not statistically significant. However, there are vast changes in expression of genes involved in immune evasion among these strains. These data suggest that the resistant parasite escapes the effective action of the drug, and this may also be due to the changes in the immune evasion genes. Thus, the whole transcriptome analysis using RNA-Seq provides extensive information about the gene transcript, from transcription start site to polyadenylation signal (Otto et al., 2010).
Conclusion
On a gene basis, much information can be gained from this study, although there is no significant information about noncoding regions, which is due to the high AT-rich content and low complexity of P. falciparum genome. The results by these data provided a basis of gene expression changes when the resistant strain was cultured in the presence of chloroquine drug. These data will help to identify potent diagnostic targets and novel candidates in the field of drug therapies for P. falciparum. Future efforts are to extend the RNA-Seq transcriptome study to all stages of the parasite culture in chloroquine condition, and also to sequence transcripts from clinical isolates to find the differences in gene expression between the drug resistances. Thus, the exhibition of transcriptional differences between these strains of P. falciparum aids to our knowledge about the drug sensitivity phenotypes and thereby promoting the current efforts for developing novel diagnostics and therapeutics for malaria.
Footnotes
Acknowledgment
This work was supported by an Institute Research Grant from Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), Puducherry, India.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.