
Abstract
Abstract
Ovarian cancer is one of the most common cancers and has a high mortality rate due to insidious symptoms and lack of robust diagnostics. A hitherto understudied concept in cancer pathogenesis may offer new avenues for innovation in ovarian cancer biomarker development. Cancer cells are characterized by an increase in network entropy, and several studies have exploited this concept to identify disease-associated gene and protein modules. We report in this study the changes in protein–protein interactions (PPIs) in ovarian cancer within a differential network (interactome) analysis framework utilizing the entropy concept and gene expression data. A compendium of six transcriptome datasets that included 140 samples from laser microdissected epithelial cells of ovarian cancer patients and 51 samples from healthy population was obtained from Gene Expression Omnibus, and the high confidence human protein interactome (31,465 interactions among 10,681 proteins) was used. The uncertainties of the up- or downregulation of PPIs in ovarian cancer were estimated through an entropy formulation utilizing combined expression levels of genes, and the interacting protein pairs with minimum uncertainty were identified. We identified 105 proteins with differential PPI patterns scattered in 11 modules, each indicating significantly affected biological pathways in ovarian cancer such as DNA repair, cell proliferation-related mechanisms, nucleoplasmic translocation of estrogen receptor, extracellular matrix degradation, and inflammation response. In conclusion, we suggest several PPIs as biomarker candidates for ovarian cancer and discuss their future biological implications as potential molecular targets for pharmaceutical development as well. In addition, network entropy analysis is a concept that deserves greater research attention for diagnostic innovation in oncology and tumor pathogenesis.
Introduction
O
Computational analysis of gene expression data has been useful for medical diagnosis and prognosis of complex human diseases, such as cancers. The ability to analyze such data at systems level, rather than conventional strategies considering individual genes, has gained importance in improving our understanding of disease-related pathways and identification of prognostic and therapeutic target gene or protein sets (i.e., system biomarkers). In this respect, biomolecular networks, such as protein–protein interactions (PPIs) (Karagoz et al., 2016) and transcriptional regulatory (Gov and Arga, 2016) and metabolic network analyses (Mardinoglu et al., 2013), constitute a scaffold for innovative data analysis so as to unravel novel disease mechanisms (Sevimoglu and Arga, 2014; Vidal et al., 2011) and to mention molecular signatures of human diseases (Calimlioglu et al., 2015; Karagoz et al., 2015; Kori et al., 2016).
Differential network analysis focuses on specific dynamic response of systems under a novel condition or disease status to elucidate the alterations in molecular mechanisms of a biological system (Ideker and Krogan, 2012). Several algorithms have been developed to analyze the topological changes in biomolecular networks under a perturbation (Gambardella et al., 2013; Zhang et al., 2016; Zickenrott et al., 2016). In addition, the efficiency of differential network analysis in elucidating the dynamic response of several cells and tissues in response to disease conditions has been reported for several human diseases, such as type 2 diabetes (Sun et al., 2013), acute myeloid and lymphoblastic leukemia (Gill et al., 2014), Alzheimer's disease (Xia et al., 2014), ovarian cancer (Hsiao et al., 2016), and breast cancer (Hsiao et al., 2016; Warsow et al., 2013).
Cancer cells are characterized by an increase in network entropy (Teschendorff et al., 2015; West et al., 2012), and this concept has been utilized in several studies to identify disease-associated gene clusters and protein interactions (Lecca and Re, 2015; Varadan and Anastassiou, 2006; Xiong et al., 2010), to quantify the level of disorder at the transcriptional level as a result of the genomic reprogramming of yeast cells under genetic perturbations (Gov and Arga, 2014), and to provide a quantitative readout of the average undifferentiated state and cellular plasticity of the cancer stem cells (Banerji et al., 2013).
We suggest in this study several PPIs as biomarker candidates for ovarian cancer using differential gene expression as a proxy. The uncertainties of the up- or downregulation of PPIs in ovarian cancer were estimated within a novel differential interactome network framework utilizing the entropy concept, a comprehensive gene expression data, and human protein interactome (Fig. 1). Following categorization of gene expression data into discrete levels, the observation frequencies of PPIs in each state were quantified within healthy and diseased groups. Then, entropies were determined and the interacting protein pairs with minimum entropies were identified. The analyses resulted with several PPIs as potential biomarkers which were scattered in several modules, each indicating significantly affected biological pathways in ovarian cancer.
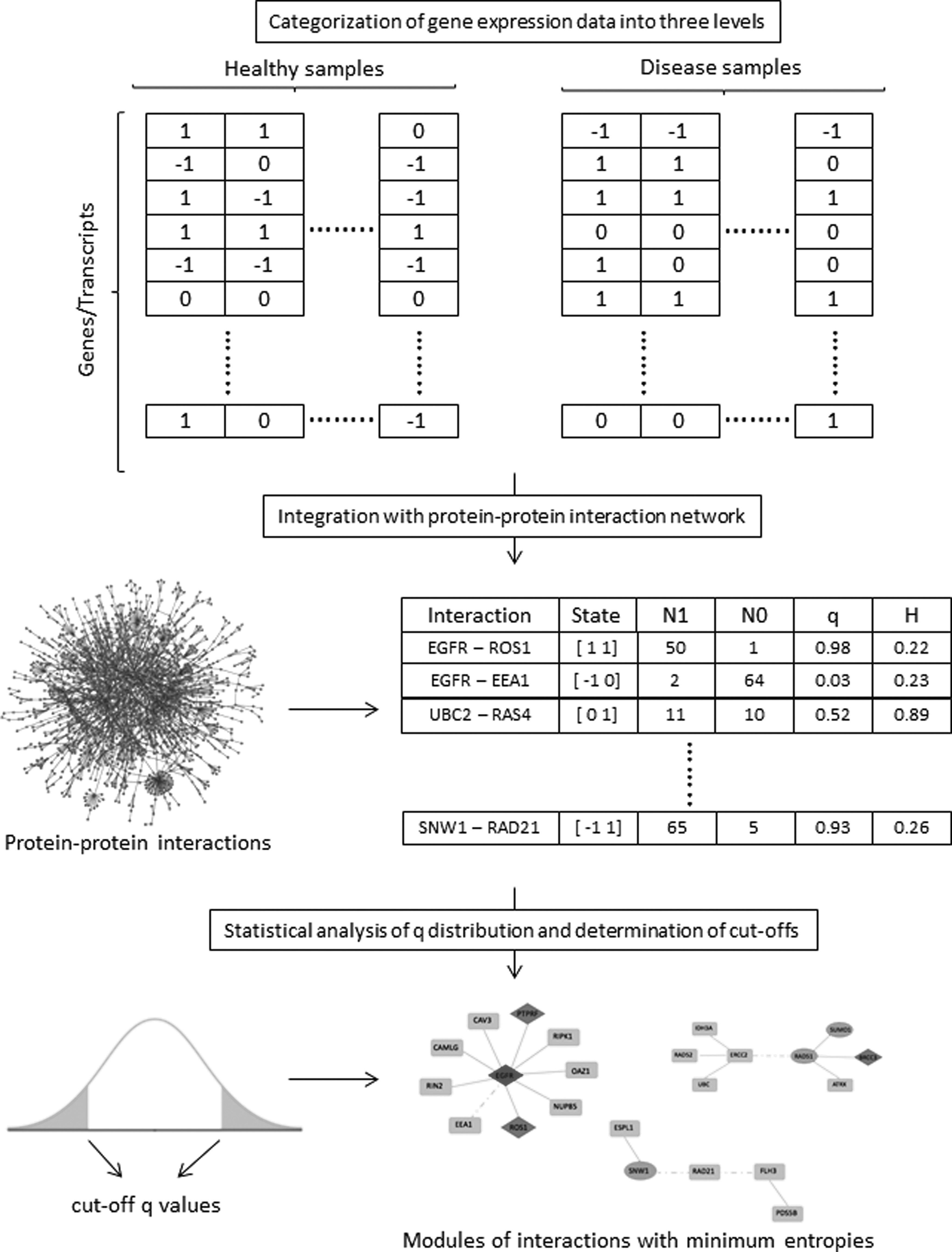
Conceptual summary of the methodology. N0 represents the number of times that the state appeared in OSE group and similarly N1 stands for the number of times that the state appeared in CEPI group. q value represents the probability that an interaction at a specified state is encountered in ovarian cancer, and H represents the corresponding entropy values. CEPI, laser microdissected epithelial samples from ovarian cancer cell; OSE, healthy ovarian surface epithelial.
Materials and Methods
Gene expression data
Taking into account the heterogeneous nature of ovarian cancer, in this study, we used a compendium of six datasets that included laser microdissected epithelial samples from ovarian cancer cell (CEPI) population and healthy ovarian surface epithelial (OSE) tissues (Table 1). The compendium consisted of 191 samples; that is, 140 CEPI and 51 OSE samples. All datasets were produced in Affymetrix platform using the Human Genome U133 Plus 2.0 arrays.
Datasets are denoted by corresponding IDs in Gene Expression Omnibus.
Protein interaction data
The human protein interactome was taken from our previous study (Karagoz et al., 2016), where confidence levels of binary PPIs were presented through incorporating eight biological features in a scoring scheme with a large group of PPIs obtained by compiling data from various human protein interaction databases. In this study, the high confidence human protein interactome with confidence score ≥0.8 was used. The interactome set consisted of 31,465 interactions among 10,681 proteins.
Determination of entropies corresponding to each interaction
We enriched the entropy minimization formulation (developed by Varadan and Anastassiou, 2006 and adapted by Xiong et al., 2010) considering several technical and statistical issues (given below) to identify the interacting protein pairs whose combined expression levels were altered (down- or upregulated) in ovarian cancer samples compared to healthy samples.
We categorized the gene expression data into three levels. Within each sample, if the expression level of a gene was significantly higher than the average, it was assigned as “1”; if the expression level of a gene was significantly lower than the average, it was assigned as “−1.” Otherwise, it was assigned as “0.” Each sample (raw .CEL microarray files) was normalized through computing the Robust Multiarray Average expression measure (Bolstad et al., 2003), and discrimination among levels was decided through employment of Student's t-test.
Benjamini–Hochberg procedure was followed to control False Discovery Rate, and adjusted p < 0.05 was used to determine statistical significance. According to the three-level formulation, 32 possible gene expression states were defined (i.e., [0 0], [0 1], [0 −1], [1 0], [1 1], [1 −1], [−1 0], [−1 1], [−1 −1]) for each interacting protein pair. For each state, we counted the number of times (N0) that the state appeared in OSE group and the number of times (N1) that it appeared in CEPI group. Considering the imbalance between the sample sizes of groups, we normalized the counting parameters taking into account the sizes of OSE (NOSE) and CEPI (NCEPI) groups (which are the maximum possible numbers of N0 and N1, respectively) and employed in calculation of q value, which represents the estimation of probability that this state is encountered in ovarian cancer:
This formulation is applied for all states that have been encountered at least once. The uncertainty of determining whether or not the state is encountered in a perturbed condition was estimated from the entropy H:
The entropy function H takes its minimum values at q values close to either 0 or 1, which represents differential observation tendency of the state between OSE and CEPI conditions, and takes a maximum value of 1 for q = 0.5 (which represents a balanced observation of the state in both conditions). To find most informative PPIs associated with ovarian cancer, we set a cutoff value for entropy H through the analysis of the distribution of q values. The distribution of q values was following normal distribution, assessed by Lilliefors corrected Kolmogorov–Smirnov test, and cutoff q values and corresponding H values (obtained through Eq. 2) were determined considering p < 0.05.
Based on q values, the following nomenclature was used to designate the status of a PPI: if the prevalence of an interaction is significantly higher in ovarian cancer (CEPI) group compared to healthy (OSE) group, the interaction was indicated as “upregulated in ovarian cancer.” Similarly, if the prevalence of an interaction is significantly higher in OSE condition with respect to CEPI condition, the interaction was indicated as “downregulated in ovarian cancer.”
Gene set enrichment analysis
Gene set enrichment analyses of gene sets were performed through DAVID bioinformatics tool (v.6.8) (Huang et al., 2009) using KEGG (Kanehisa et al., 2016) as the data source for molecular pathways and Gene Ontology (Ashburner et al., 2000) as the data source for biological processes. Whole genome annotation for human genome was used as the background reference set. A modified Fisher Exact test was used to determine p-values (Huang et al., 2009), and p < 0.05 was used to determine the statistical significance of enrichment results. In addition, to characterize the molecular functions of each gene/protein and their association with diseases, we manually searched GeneCards Human Gene Database (Fishilevich et al., 2016).
Results
In the present study, to estimate the uncertainties of the up- or downregulation of PPIs in ovarian cancer and to predict PPIs as biomarker candidates, we used gene expression levels as a proxy. For ideal interpretation of transcriptome data, we used a compendium of transcriptome datasets, including CEPI and OSE cells, which resulted with an extensive dataset, including 140 CEPI and 51 OSE samples (Table 1). The gene expression data were categorized into three levels, and the prevalence of PPIs in CEPI and OSE samples was determined. The q values, which represent the estimation of probabilities that each PPI in a certain state is encountered in ovarian cancer, were normally distributed (Lilliefors corrected Kolmogorov–Smirnov test with p > 0.80) with an average and standard deviation of 0.46 and 0.15, respectively (Fig. 2). Considering a 99.5% confidence interval (i.e., p < 0.05), q values >0.85 or <0.07 and corresponding H values (obtained through Eq. 2), which are <0.38, were considered as significant. As a result, a total of 1216 PPIs were recorded as significant (Supplementary Table S1).

The distribution of q values, representing the probability that an interaction at a specified state is encountered in ovarian cancer.
Following our nomenclature (described in Materials and Methods section), we observed that 22 PPIs were upregulated in ovarian cancer, while 1194 PPIs were downregulated. Furthermore, differential PPI patterns were observed around 105 proteins, displaying several upregulated interactions besides downregulated PPIs. These proteins (and their interactions) were analyzed further to assess molecular aspects of cellular responses in ovarian cancer.
We observed that the differential PPIs belonging to 105 proteins were grouped into 11 modules (Fig. 3). Through functional enrichment analyses, the modules were related with specific pathways and biological processes. Significant associations were determined for main pathways of DNA repair mechanisms (namely, the nucleotide-excision repair [NER], homologous recombination [HR], and nonhomologous end joining [NHEJ]), alternative splicing mechanism, proliferation-related mechanisms, nucleoplasmic location of estrogen receptor, interactions of MYC proto-oncogene, extracellular matrix (ECM) degradation, and inflammation response.
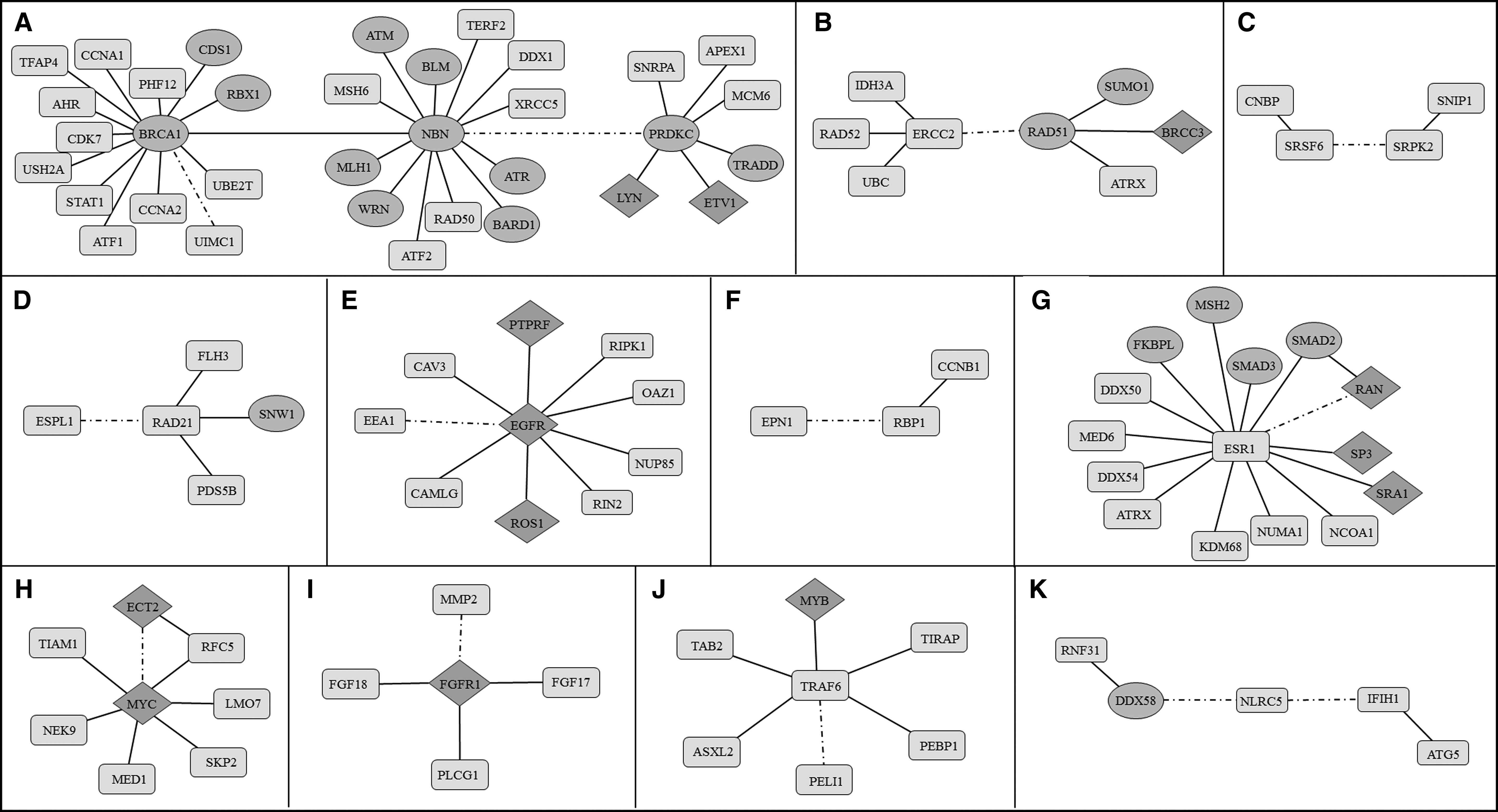
Eleven modules (
In the present study, two modules were associated with DNA damage (p = 8.1 × 10−8) and repair mechanisms (p = 5.1 × 10−10) in ovarian cancer (Fig. 3A, B). Among the five major repair mechanisms, HR, NHEJ, and NER were represented by these modules. The major module (Fig. 3A) consisting of 34 proteins included 12 tumor suppressor proteins (ATM, ATR, BARD1, BLM, BRCA1, CDS1, MLH1, NBN, PRKDC, RBX1, TRADD, and WRN) and 2 proteins encoded by oncogenes (ETV1 and LYN). At the center of this module, three antitumor proteins exist: BRCA1, NBN, and PRKDC. Second module (Fig. 3B) consisted of 8 proteins, among which RAD51 and SUMO1 are tumor suppressors, whereas BRCC3 is an oncogene. RAD51 and ERCC2 were the central proteins of this module.
The third module consisting of four proteins (Fig. 3C), that is, CNBP, SRSF6, SRPK2, and SNIP1, pointed out the regulatory role of alternative splicing mechanisms (p = 0.0125) in ovarian cancer. An important mechanism in ovarian cancer proliferation through sister chromatid separation (p = 3.4 × 10−4) was represented through the module around RAD21 (Fig. 3D). The interaction between RAD21 and ESPL1 was upregulated in ovarian cancer.
The stress response (p = 1.6 × 10−4) module around epidermal growth factor receptor, EGFR, consists of interactions with two oncogenes, PTPRF and ROS1, as well as CAV3, CAMLG, NUP85, RIN2, RIPK1, and OAZ1, which were downregulated in ovarian cancer (Fig. 3E). In addition, the interaction of EGFR with the early endosomal antigen 1 (EEA1) was upregulated, indicating the transportation of EGFR through EEA1 positive endosomes in ovarian cancer (Oksvold et al., 2000). The interaction of retinol binding protein 1 (RBP1) with Epsin 1 (EPN1) was also upregulated in ovarian cancer (Fig. 3F) to suppress the EGFR-associated proliferation through retinoids.
The module around estrogen receptor 1 (ESR1) consists of interactions with several proteins, including four oncogenes (SP1, SP3, SRA1, and RAN) and four tumor suppressors (FKBPL, MSH2, SMAD2, and SMAD3), and was enriched with positive regulation of transcription (p = 6.6 × 10−6). The interaction between ESR1 and RAN required special attention, since it was upregulated in ovarian cancer in contrast to the other interactions in the module (Fig. 3G).
The module around MYC protein consists of several interactions in regulation of signaling (p = 2.2 × 10−4) and protein modification process (p = 7.7 × 10−4); however, the interaction between MYC and Epithelial Cell Transforming 2 (ECT2) proteins needs more attention since these are both proto-oncogenes and their interaction was upregulated in ovarian cancer unlike the other interactions in the module (Fig. 3H).
The module representing the transmembrane receptor protein tyrosine kinase signaling pathway (p = 2.2 × 10−6) and the regulation of endothelial cell migration (p = 9.6 × 10−5) has Fibroblast Growth Factor Receptor 1 (FGFR1) protein in the center, and its interaction with Matrix metalloproteinase 2 (MMP2) in ovarian cancer was emphasized (Fig. 3I). The module, which appears to be working on the activation of inflammatory response (p = 3.8 × 10−5), was composed of several proteins around TRAF6. Interaction of TRAF6 with PELI1 was upregulated in ovarian cancer (Fig. 3J). The other immune response (p = 7.3 × 10−5) module around NOD-Like Receptor C5 (NLRC5) included upregulated interactions with DDX58 and IFH1 proteins in ovarian cancer (Fig. 3K). In addition, while studying on the modules, we recognized that these modules were actually communicating with each other, and it was possible to represent the response of the cells in a single PPI network with addition of one or two proteins linking two modules (Fig. 4). A few proteins (namely, AKT, BRCC3, FLH3, and TGF1) making the connection between these modules were not detected in our analysis that is why the whole network looks as an individual module.
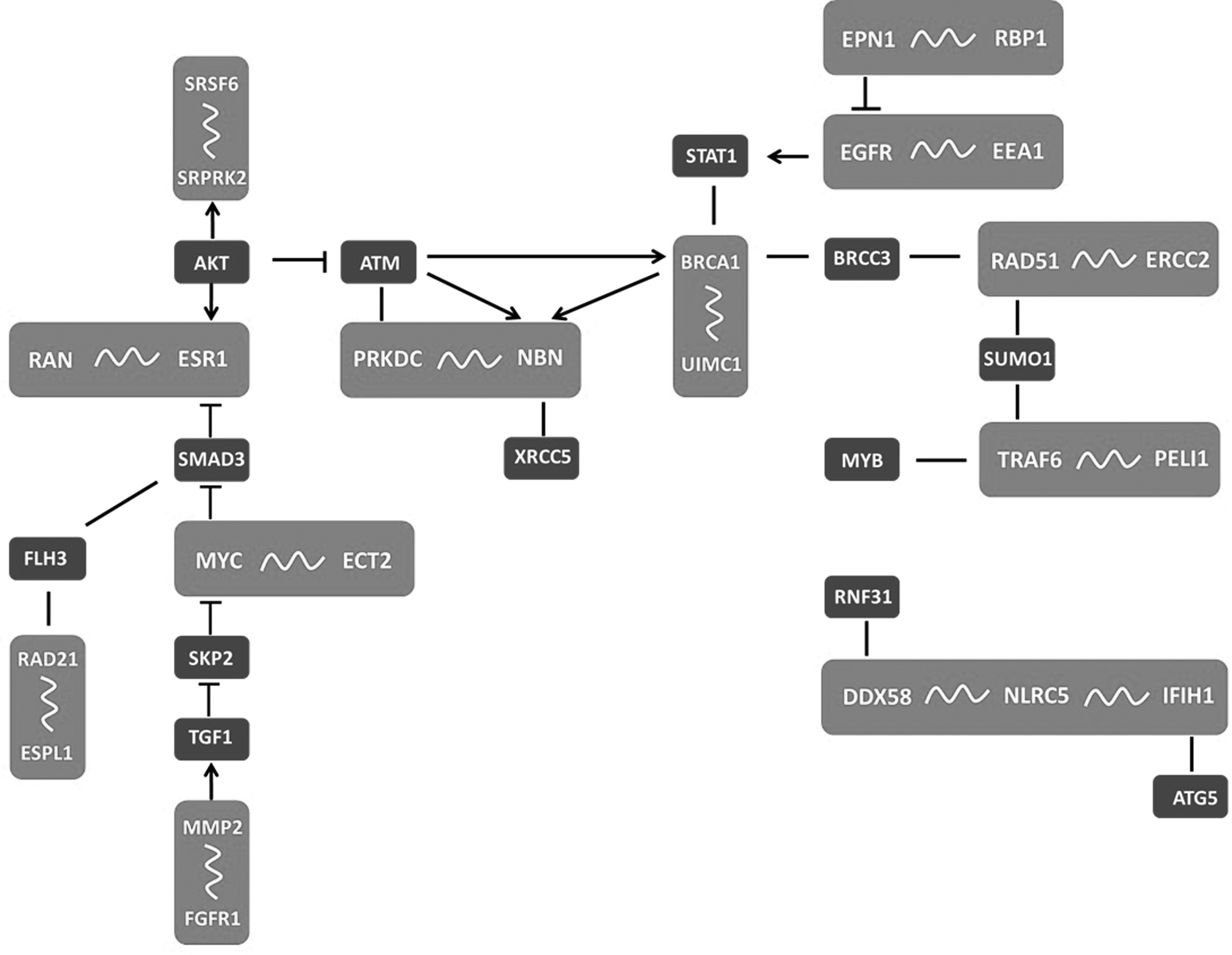
Response map indicating communication among 11 separate modules in ovarian cancer. Wavy lines and straight lines indicate interactions upregulated and downregulated interactions, respectively. Arrows indicate activation, blunt ends represent suppression mechanisms.
Discussion
Elucidation of the molecular aspects of cellular response in complex diseases, such as cancers, is a great challenge. System biomedicine approaches, integrating high-throughput functional genomics data with genome-scale biomolecular networks, offer potential solutions at molecular level and pave the way for understanding molecular pathogenesis of the diseases, identification of effective biomarkers for early diagnosis, and development of effective treatment strategies. We report in this study the changes in PPIs in ovarian cancer within a differential network (interactome) analysis framework utilizing the entropy concept, a comprehensive gene expression data, and human protein interactome. The salient findings and their broader integrative biology context or relevance to biomarker and diagnostics innovation in cancers are discussed below.
To estimate the up- or downregulation of PPIs in ovarian cancer, we in this study used gene expression levels as a proxy. To increase dimensionality and breadth of information that can be extracted, we did not limit the analyses with a single transcriptomics dataset, but constructed a compendium of various datasets. In addition, a single type of microarray was chosen as the investigation platform to reduce the confounding factors in the data analysis, such as variabilities originated from array platforms. Moreover, considering the cellular complexity of tumor tissues, we analyzed laser microdissected epithelial samples only.
The entropy based analyses performed in this study resulted with significant alterations in 1216 PPIs, the majority of which were downregulated in ovarian cancer. Differential PPI patterns were accumulated around 105 proteins clustered in 11 modules, depicting the differences between healthy and disease conditions in the regulation of several signaling and developmental processes, including DNA repair, cell proliferation-related mechanisms, nucleoplasmic translocation of estrogen receptor, ECM degradation, and inflammation response. Then, with the inclusion of a few proteins making the connection between the individual modules, it has become possible to represent the response of the cells in a single PPI network. Indeed, this functional PPI network showed how the complexity of the cancer is related with the key proteins and their physical interactions like domino tiles regarding different biological mechanisms.
Based on the comprehensive literature survey on the relevance of resultant proteins and their interactions in ovarian cancer, we suggest several PPIs as biomarker candidates for ovarian cancer. First, all the interactions of DNA repair module were downregulated in ovarian cancer with the exception of these three interactions: BRCA1 and UIMC1, NBN and PRKDC, and ERCC2 and RAD51, which were upregulated. It was determined that these interactions were related with the cellular response for the detection of DNA damages and stimulation of DNA repair pathways in ovarian cancer (Stoppa-Lyonnet, 2016).
The importance of the interaction between SRSF6 and SRPK2 detected in our analysis is in accordance with the literature stating that the alternative splicing mechanisms and abnormal protein expression are key processes in tumor cells (Ritchie et al., 2008). Thus, interaction between the splicing protein SRSF6 and its kinase protein SRPK2 might be another potential biomarker due to mimicking the aberrant expression of splicing factors, frequently observed in cancers.
Separation of sister chromatids and aneuploidy are known as hallmarks for human malignancies (Duesberg et al., 2006). In this concept, the results of the analysis pointed out the interaction between ESPL1 and RAD21 as a potential target since this interaction during the cleavage of cohesion protein complex causes chromosomal instability and aneuploidy in human cancer cells due to improper chromosome segregation (Solomon et al., 2011).
The interaction between EPN1 and RBP1 has taken attention because of their roles in the suppression of EGFR-associated proliferation through EGFR endocytosis and retinoids in ovarian cancer (McMahon and Boucrot, 2011; Tessneer et al., 2013). Furthermore, loss of function of the EPN1 gene was associated with reduced tumor growth and progression in certain cancer types (Tessneer et al., 2013).
Kau et al. (2004) reported that the alterations in nuclear transport could cause abnormal nuclear-cytoplasmic transport of oncogenes and tumor suppressors, and these molecules can be mislocalized in cancer cells. The interaction between ESR1 and RAN proteins might be considered as potential drug target in therapeutics of ovarian cancer considering their findings since this interaction mediates the nucleocytoplasmic translocation of estrogen receptor.
Rest of the modules has represented information about response of the cells to malignancies. The upregulated interaction between these two oncogenes, MYC and ECT2, may enhance the overexpression of ECT2 leading to malignant transformation of the cells (Huff et al., 2013; Kim et al., 2005). The interaction between FGFR1 and MMP2, which triggers ECM degradation, could be related with lots of processes, tumor invasion, metastasis, and angiogenesis (Komatsu et al., 2004). As expected, inflammatory response of malignant cells was determined. The interaction between TRAF6 and PELI1 forms a complex with interleukin-1 receptor associated kinase 1 (IRAK1), which triggers induction of the NF-kB signaling pathway initiating inflammatory response (Jensen et al., 2003). It may suggest that the interactions of NLRC5 with RNA helicases, DDX58 and IFIH1, may block the inflammatory response to abnormal accumulation of ribonucleic acids formed in tumorigenesis (Cui et al., 2010). Hence, the growth arrest and cell death in exposed cell populations could not be possible because of the inhibition of inflammatory response to this ribonucleic acid accumulation. In this study, we propose that NLRC5 could be a possible drug target for arresting tumor cell growth and initiating cell death.
In summary, all the aforementioned interactions could be considered as potential biomarkers for diagnostic innovation in ovarian cancer. Furthermore, the following interactions were also proposed as potential drug targets in ovarian cancer therapeutics: (1) The interaction between SRSF6 and SRPK2, mimicking the aberrant expression of splicing factors (Ritchie et al., 2008). SRPK2 inhibitors such as adenine (Vitamin B4), purvalanol, and phosphoaminophosphonic acid-adenylate ester could be considered as potential drugs. (2) The interaction between ESPL1 and RAD21 might be blocked to suppress RAD21 activity and to avoid chromosomal instability and aneuploidy due to improper chromosome segregation as a result of cleavage of cohesion protein complex (Solomon et al., 2011). (3) The interaction between EPN1 and EEA1 could be considered as a direct target for EPN1 inactivation to reduce tumor growth and progression (Tessneer et al., 2013). (4) The interaction between ESR1 and RAN, which results in nucleocytoplasmic translocation of estrogen receptor in ovarian cancer (Kau et al., 2004). (5)The interactions of NLRC5 with DDX58 and IFIH1 to arrest tumor cell growth and initiate cell death (Cui et al., 2010).
Network based analyses within the systems biological point of view deserved the attention depending on its high potential for gaining wider biological perspective in diagnostic and therapeutic researches. However, integration of the entropy concept within a differential network analysis framework in the present study is taking this approach to a higher level in terms of taking into consideration the dynamics of the interactions, as well as quantifying the uncertainties in predictions. By this means the approach enables to predict the physical interactions between proteins as potential biomarkers (and also drug targets) unlike the prevalent proposals in development of diagnostic tests, which are based on gene and protein expression levels. From this perspective, network entropy analysis is a concept that deserves greater research attention for diagnostics innovation in oncology and tumor pathogenesis. In addition, the used methodology not only uses the differentially expressed genes like the conventional approach but also uses the expression patterns of the whole transcripts. Thereby, it is possible to catch up the PPIs that may have vital roles in disease conditions, although the expressions of genes encoding these interacting proteins may not be significantly higher or lower than the population average.
Conclusions and Future Outlook
A novel system's biomedicine strategy utilizing the network entropy concept and differential network analysis framework was used together to enlighten the response of epithelial cells in ovarian cancer. One hundred five proteins with changing PPI patterns among healthy and disease conditions were found scattered in 11 modules each indicating the significantly affected biological pathways to give a clue about potential biomarkers and drug targets for the further analyses.
In the present study, significant associations in ovarian cancer were determined as DNA repair mechanisms (i.e., NER, HR, and NHEJ), activation of alternative splicing mechanisms, proliferation-related mechanisms, nucleoplasmic translocation of estrogen receptor, interaction of MYC proto-oncogene, ECM degradation, and inflammation response. In this study, we propose several PPIs as candidate biomarkers and potential drug targets, which could be considered for improving diagnostic tools and developing treatment strategies in further experimental and clinical studies.
Footnotes
Acknowledgments
Support by Marmara University Scientific Research Projects Committee (BAPKO) in the context of the project FEN-C-DRP-110915-0445.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.