
Abstract
Abstract
Biological psychiatry research has long focused on the brain in elucidating the neurobiological mechanisms of anxiety- and trauma-related disorders. This review challenges this assumption and suggests that the gut microbiome and its interactome also deserve attention to understand brain disorders and develop innovative treatments and diagnostics in the 21st century. The recent, in-depth characterization of the human microbiome spurred a paradigm shift in human health and disease. Animal models strongly suggest a role for the gut microbiome in anxiety- and trauma-related disorders. The microbiota–gut–brain (MGB) axis sits at the epicenter of this new approach to mental health. The microbiome plays an important role in the programming of the hypothalamic–pituitary–adrenal (HPA) axis early in life, and stress reactivity over the life span. In this review, we highlight emerging findings of microbiome research in psychiatric disorders, focusing on anxiety- and trauma-related disorders specifically, and discuss the gut microbiome as a potential therapeutic target. 16S rRNA sequencing has enabled researchers to investigate and compare microbial composition between individuals. The functional microbiome can be studied using methods involving metagenomics, metatranscriptomics, metaproteomics, and metabolomics, as discussed in the present review. Other factors that shape the gut microbiome should be considered to obtain a holistic view of the factors at play in the complex interactome linked to the MGB. In all, we underscore the importance of microbiome science, and gut microbiota in particular, as emerging critical players in mental illness and maintenance of mental health. This new frontier of biological psychiatry and postgenomic medicine should be embraced by the mental health community as it plays an ever-increasing transformative role in integrative and holistic health research in the next decade.
Introduction
T
The Human Gut Microbiome
“Human microbiota” is the term used to describe all the microorganisms (bacteria, eukaryotes, archaea, and viruses) within the human body, while the microbiome is defined as the complete catalog of these microbes and their genes (Dave et al., 2012). The numbers of bacterial and human cells are estimated to be close to equal, with about 3.9 × 1013 bacterial cells and 3.0 × 1013 human cells (Sender et al., 2016). Research has shown that the composition of the microbiota changes across the life span (Douglas-Escobar et al., 2013).
It was originally believed that the intestines were sterile in utero, however, recent evidence suggests that there is a degree of maternal–fetal bacterial transmission via the amniotic fluid and/or umbilical cord blood (Al-Asmakh et al., 2012; Jiménez et al., 2005; Satokari et al., 2009; Wagner et al., 2008) and bacterial species have been detected in the meconium of healthy neonates (Jiménez et al., 2008). During and immediately following delivery, the newborn is exposed to the microbiota of the mother as well as the environment to acquire a range of commensal intestinal bacteria (Hooper et al., 2012).
During the perinatal period, the functional development of the mammalian brain is susceptible to both internal and external environmental cues. Epidemiological studies have found an association between microbial pathogen infections during this period and common neurodevelopmental disorders, such as autism and schizophrenia (Finegold et al., 2002; Mittal et al., 2008). Similarly, exposure to microbial pathogens in rodents during developmental periods results in, among others, anxiety-like behaviors and impaired cognitive function (Bilbo et al., 2005; Goehler et al., 2008; Sullivan et al., 2006). Desbonnet et al. (2010) showed that the commensal bacterium, Bifidobacterium infantis, has the ability to modulate tryptophan metabolism, suggesting that the gut microbiota can influence the precursor pool for serotonin and various other bioactive tryptophan metabolites.
These results underscore the importance of the gut microbiome in very early-life stages and its effects on neurodevelopment and mental health. It could also provide insights into the observed associations between early-life trauma and susceptibility to the development of anxiety- and trauma-related disorders later in life (Famularo et al., 1992; Felitti et al., 1998; McCauley et al., 1997).
Over the past few years, a new research field has emerged that investigates the human microbiome with the goal of determining how the composition of the gut microbiome influences health and disease. This has given rise to large international collaborative projects, such as the Human Microbiome Project (HMP) (Human Microbiome Project Consortium, 2012) and MetaHIT (Qin et al., 2010). A study conducted by the HMP detected marked interindividual differences in the microbiota of healthy controls. Metabolic pathways were, however, stable among individuals, despite variation in community structure. Furthermore, ethnic/racial background was one of the strongest associations of both microbes and pathways with clinical metadata (Human Microbiome Project Consortium, 2012).
Two large-scale studies of thousands of healthy individuals, the Belgian Flemish Gut Flora Project and the Dutch LifeLines-DEEP study, found that gut microbiome composition correlated with several factors, including stool consistency, diet, use of medication, red blood cell counts, and fecal chromogranin A (Falony et al., 2016; Zhernakova et al., 2016). No associations with microbiota composition variation and mode of delivery, infant feeding, and residence type were found. The authors noted that the lack of signal in the data was unexpected, and that their results do not imply that early-life events do not affect microbiota assembly during infancy, but only indicate that these events are not significantly associated with microbiome composition in adulthood in those cohorts [see review by Tamburini et al. (2016) for discussion of factors that influence microbial homeostasis during early life that were associated with the development or protection against disease during childhood].
The authors also made recommendations for sample size (power) determination, emphasizing the importance of large-scale microbiome studies and the inclusion of known covariates to detect shifts in microbial composition (Falony et al., 2016).
The MGB Axis
The bidirectional communication between the brain and gut microbiota has been termed the MGB axis, and preclinical studies indicate that dysbiosis (dysregulation of the microbiota) influences anxiety and stress behaviors (Foster and McVey Neufeld, 2013), suggesting that the MGB could influence the risk of disease, including anxiety and mood disorders. The bidirectional interactions between the gut microbiota and critical parts of the central nervous system (CNS) and immune systems are maintained through direct and indirect pathways, which include endocrine (hypothalamic–pituitary–adrenal [HPA] axis) (Sudo et al., 2004), immune (chemokines, cytokines) (Macpherson and Harris, 2004), and metabolic pathways (Diaz-Anzaldua et al., 2011), the limbic system (Carabotti et al., 2015), as well as the efferent (Rao and Gershon, 2016; Liu et al., 2009), afferent (Wood, 2008), and sympathetic afferent systems (Mayer, 2011).
Communication between the visceral afferent, limbic, and autonomic systems provides the neural connections that underlie the link between behavior and gut function in health and disease (Mayer, 2011). In addition, several other factors may also impact the MGB, including the HPA axis, neurotransmitters produced by the gut microbiota [such as tryptophan and serotonin; reviewed by O'Mahony et al. (2015)], and the integrity of the brain/blood barrier (BBB) (Braniste et al., 2014) and intestinal epithelial barrier (Söderholm et al., 2002).
The Gut Microbiome in Anxiety and Stress
The majority of earlier microbiome studies focused on animal models, which are convenient model systems that provide increased control over genetic and environmental factors that influence the microbiome. The germ-free (GF) animal model is a powerful tool to examine the effects of the microbiota on behavior in an attempt to determine causation and to study the effect of particular bacteria or a dietary intervention on the MGB axis. GF animals exhibit major alterations in gastrointestinal and immune system functioning, which could have important effects on the brain and behavior (Crumeyrolle-Arias et al., 2014; Sudo et al., 2004). However, it should be emphasized that this relationship is also influenced by temporal, strain, sex, and species factors (Mayer et al., 2014), all of which are not yet fully understood.
To show that the microbiota can directly affect behavior, researchers transplanted microbiota from adult GF BALB/c mice (a high-anxiety mouse strain) into adult GF NIH Swiss mice (a low-anxiety mouse strain), and the BALB/c mice received the microbiota of the NIH Swiss mice. Following fecal transplantation, the behavioral profile of the donor was evident in the recipient animal (Bercik et al., 2011a). While it was originally suggested that the critical window for recolonization to reverse the anxiolytic phenotype is during early-life/adolescence (Clarke et al., 2013; Neufeld et al., 2011; Stilling et al., 2014), this study and a few others (Collins et al., 2012; Nishino et al., 2013) have illustrated that the behavior of GF animals is susceptible to modification even during adulthood. The behavior of GF animals is quite distinct from that exhibited by control animals. Sudo et al. (2004) discovered that GF mice exhibited an exaggerated HPA axis response to restraint stress, which was reversed following monocolonization with a particular Bifidobacterium species.
Similarly, when comparing GF and specific pathogen free (SPF) stress-sensitive, F344 rats, GF rats showed exaggerated neuroendocrine responses and increased anxiety-like behavior compared to SPF animals (Crumeyrolle-Arias et al., 2014). Another study found that short-term colonization of GF mice in adulthood is able to reduce anxiety-like behaviors (Nishino et al., 2013). Moreover, variations in neurotransmitter signaling have also been observed in specific brain regions of GF mice (Diaz Heijtz et al., 2011) as well as altered HPA axis functioning (Sudo et al., 2004).
The relationship between the microbiome and behavior is, however, not unidirectional; stress and emotions can influence the gut microbial composition through the release of stress hormones or sympathetic neurotransmitters that influence gut physiology and alter the habitat of the microbiota (Montiel-Castro et al., 2013). Preclinical studies have shown that psychological stress, including maternal separation and restraint, heat, and acoustic stress, alters the composition of the gut microbiota (Bailey et al., 2011; De Palma et al., 2014; Moloney et al., 2014). Furthermore, stress has the ability to increase intestinal permeability, probably through the involvement of corticotrophin releasing factor (CRF) and its receptors (CRFR1 and CRFR2), which play a key role in stress-induced gut permeability dysfunction (Overman et al., 2012; Rodiño-Janeiro et al., 2015; Taché and Million, 2015).
Increased intestinal permeability provides bacteria an opportunity to translocate across the intestinal mucosa and directly access both the immune and neuronal cells of the enteric nervous system (ENS) (Gareau et al., 2008; Teitelbaum et al., 2008). Stress also activates the autonomic nervous system, which affects gastric acid, bile, and mucus secretion, as well as gut motility (Beckh and Arnold, 1991; Shigeshiro et al., 2012; Söderholm and Perdue, 2001). Gut motility is of particular importance since it is strongly associated with gut microbiota composition and richness, and it is therefore important to capture this information in microbiota studies (Falony et al., 2016; Vandeputte et al., 2016).
Preclinical microbiome data have been extrapolated to humans, and although there has been an exponential increase in the number of human microbiome studies in general, there is still an underrepresentation of microbiome research in psychiatric disorders. The majority of clinical studies focus on key microorganisms as potential psychobiotics (microorganisms that exhibit positive effects on the CNS) in healthy (Kelly et al., 2017) or affected individuals (refer to The Gut Microbiome as a Therapeutic Target section).
Uncontrolled inflammatory responses are evident in patients with PTSD and have been shown to play a role in the pathogenesis of the disorder. Altered regulatory T cells (Tregs), cells that assist in maintaining optimum immune regulation and protect against inappropriate inflammatory responses, have been reported in individuals with PTSD (Morath et al., 2014; Sommershof et al., 2009). In addition, upregulated proinflammatory cytokine profiles, such as tumor necrosis factor (TNF), interferon gamma (IFN-γ), and interleukin 1 beta (IL-1β), have been observed in PTSD (Hoge et al., 2009; Lindqvist et al., 2014; Maes et al., 1999). The origins of this altered immune regulation is a point of discussion and one of the probable candidates is the human microbiome, since it is an important determinant of immunoregulation (Rook et al., 2014; Sefik et al., 2015).
In addition, the microbiota produce and utilize neuro- and immune-active substances, such as γ-aminobutyric acid, melatonin, acetylcholine, catecholamines, histamine, and serotonin, which can penetrate the gut mucosa, enter the bloodstream, and subsequently cross the BBB, and ultimately affect functioning within the CNS (Barrett et al., 2012; Theoharides et al., 2004).
The aforementioned findings lay the ground for a recent pilot study that investigated the gut microbiome in PTSD patients (Hemmings et al. in press). Although the authors did not detect any differences in overall diversity measures between the PTSD patients and trauma-exposed (TE) controls, random forest analysis showed that decreased relative abundance of Actinobacteria, Lentisphaerae, and Verrucomicrobia phyla in PTSD subjects was able to distinguish PTSD from TE controls with a high degree of accuracy. Their findings were consistent with an animal model of PTSD that hypothesized that decreased exposure to Actinobacteria and other immunoregulatory/anti-inflammatory “Old Friends” could lead to increased vulnerability to PTSD (Reber et al., 2016).
Furthermore, the authors did not detect any differences in plasma C-reactive protein (CRP) concentrations between PTSD and TE controls in this small pilot study, which contrasts with earlier findings. However, the mean time since index trauma was 11 years, and other studies mostly investigated inflammatory markers at the time of trauma exposure and used healthy as opposed to TE controls. This study also had a limited sample size (18 PTSD patients and 12 TE controls) and these findings await replication in considerably larger samples that include healthy controls.
Several studies have investigated the fecal microbiota of individuals with depression and they have yielded conflicting results, both in abundances of specific taxa and in terms of diversity indexes. One study found increased bacterial diversity in depressed individuals (Jiang et al., 2015), while Kelly et al. (2017) detected lower diversity in patients with depression, and neither Naseribafrouei et al. (2014) nor Zheng et al. (2016) found significant differences in bacterial diversity between depressed individuals and healthy controls. These findings highlight the importance of taking confounding factors (such as medication, diet, and stool consistency) into account in microbiota studies (Falony et al., 2016), as these factors could explain the conflicting results.
Furthermore, much larger and well-characterized cohorts are required to establish the true relationship between the gut microbiota and depression. Although these studies did not investigate individuals with anxiety disorders, major depression is highly comorbid with anxiety- and trauma-related disorders (Elhai et al., 2008; Rytwinski et al., 2013), hence studies investigating whether similar trends exist in anxiety- and trauma-related disorders (with and without comorbid depression) are needed.
The Gut Microbiome as a Therapeutic Target
In light of the vital role of the MGB axis in CNS functioning, strategies aimed at modulating the MGB axis offer an attractive means of improving mental health outcomes. Probiotics (live, beneficial microorganisms) (Shah, 2007), prebiotics (nondigestible food substances) (Parvez et al., 2006), and synbiotics (combination of probiotics and prebiotics) (Underwood et al., 2009; Vlieger et al., 2009) have been used in attempts to modulate the gut microbiome content. The mechanisms through which probiotics potentially mediate health benefits are extensive and include several interconnected networks, including pathogen displacement (Collado et al., 2008), competition with hostile bacteria for metabolic interactions (Martin et al., 2010), production of bacteriocins (Corr et al., 2007), inhibition of bacterial translocation (Generoso et al., 2010), enhancement of mucosal barrier function (Liu et al., 2011), effects on calcium-dependent potassium channels in intestinal sensory neurons (Kunze et al., 2009), induction of cannabinoid and opioid receptors in intestinal epithelial cells (IEC) (Rousseaux et al., 2007), and modulation of the immune system (Sanders, 2011).
Preclinical investigations
Several probiotic therapies have been studied in animal models, with mostly Bifidobacterium and Lactobacillus genera eliciting beneficial effects on anxiety- and depression-like behaviors (Barrett et al., 2012; Schousboe and Waagepetersen, 2007), however, only certain strains have shown positive effects (Dinan et al., 2013). Chronic Bifidobacterium infantis treatment reduced immune alterations, depressive-like behavior, and restored noradrenaline concentrations in the brainstem in a model of early-life stress (Desbonnet et al., 2010). Lactobacillus helveticus ROO52 improved anxiety-like behavior and memory dysfunction in naive mice and mice on a western-style diet (fat 33%, refined carbohydrate 49%) (Ohland et al., 2013). Lactobacillus rhamnosus JB-1 reduced anxiety- and depressive-like behaviors and induced region-specific alterations in GABAB1b mRNA in the brain (Bravo et al., 2011) and Bifidobacterium longum effectively normalized anxiety-like behavior in a colitis model (Bercik et al., 2011b).
In addition, a strain of B. longum, but not L. rhamnosus, normalized anxiety-like behavior and levels of hippocampal brain-derived neurotrophic factor (BDNF) induced by Trichuris muris infection (nematode that causes inflammation and the appearance of anxiety-like behaviors) (Bercik et al., 2010).
A combined treatment of L. rhamnosus and L. helveticus reversed stress-induced memory dysfunction in mice infected with Citrobacter rodentium (Gareau et al., 2011). Another study found that L. plantarum treatment significantly reduced anxiety-related behavior and altered serotonergic and GABAergic neural signaling in an adult zebrafish model. Serum cortisol and leukocyte patterning revealed that supplementation with L. plantarum protected against stress-induced dysbiosis (Davis et al., 2016).
The aforementioned studies illustrate the efficacy of probiotics in mediating changes in the CNS and behavior, and underscore the strain-specific effects of probiotics. There is, however, a paucity of preclinical and clinical studies investigating the effects of prebiotics in anxiety- and stress-related behaviors. One study investigated the effects of a 5-week treatment of the prebiotic compounds fructooligosaccharide (FOS) and galactooligosaccharide (GOS) (shown to increase relative abundance of microorganisms that are thought to be beneficial) (Davis et al., 2011; Thompson et al., 2017) on the levels of BDNF and N-methyl-D-aspartate receptors (NMDARs) in rat brain (Savignac et al., 2013). Prebiotic treatment resulted in increased BDNF and NR1 subunit expression in the hippocampus. GOS treatment resulted in increased hippocampal NR2A subunits, frontal cortex NR1, and d-serine, as well as plasma d-alanine.
The authors concluded that prebiotic treatment probably altered the gut microbiota composition, facilitating increased BDNF expression in the brain, possibly through the involvement of gut hormones. GOS also elicited a stronger effect on central NMDAR signaling than FOS, suggesting a stronger proliferative potency of GOS on the microbiota (Savignac et al., 2013).
Tarr et al. investigated whether prebiotic oligosaccharides (naturally found in human milk) can inhibit stress-induced changes in gut microbial composition and attenuate stress-induced anxiety-like behavior. Exposure to the stressor resulted in anxiety-like behavior, reduction in immature neurons in the dentate gyrus, and altered colonic mucosa-associated microbiota in mice on a standard laboratory diet. None of these effects was noted in animals that were fed milk oligosaccharides. These results support the potential role of prebiotics, potentially through effects on the MGB axis, to support normal microbial functions and regulate behavioral responses in the context of a stressor (Tarr et al., 2015).
Another way to alter the composition of the gut microbiota is through dietary changes. One study showed that including 50% lean beef into normal chow significantly affected fecal bacteria composition, compared to mice on a regular chow diet (Li et al., 2009). Furthermore, this altered diet and the associated change in microbiota resulted in improved cognitive parameters and reduced anxiety-like behaviors (Li et al., 2009), suggesting that dietary interventions have the ability to alter intestinal microbiota, which could promote beneficial changes to cognitive abilities.
The field of nutrigenomics has yielded valuable information on correlations between an individual's genetic composition (host genetics) and dietary intake and how nutrition influences gene expression of the host (Pavlidis et al., 2015, 2016) and in the era of metagenomics, nutri-metagenomic approaches can be applied to unravel the interaction between the microbiota, nutrition, and host in the context of disease and as a therapeutic target (Dimitrov et al., 2016; Ferguson et al., 2016).
Organisms present in the environment can also alter homeostatic function and behavior in the host. Environmental bacteria are nonpathogenic microorganisms that inhabit our surroundings (Rook and Brunet, 2005). Some of these bacteria also form part of the “old-friends” or hygiene hypothesis, originally described by Rook et al. (2003), who proposed that reduced exposure to these “old friends” could contribute to increased immunoregulatory disorders in individuals with a suboptimum regulation of Tregs (see the Environmental Microbiome section). One such environmental bacterium is Mycobacterium vaccae, a nonpathogenic aerobic soil bacterium found in temperate environments and regarded as transient commensal (i.e., cannot colonize the digestive tract) (Gomez et al., 2001). Administration of heat-killed M. vaccae effectively downregulates symptoms of allergic inflammation through increased production of IL-10 and IFN-γ by mesenteric lymph node cells and splenocytes (Hunt et al., 2005). Lowry et al. (2007) showed administration of heat-killed M. vaccae antigen in mice activated serotonergic neurons in the dorsal raphe nucleus (DR) of the brainstem.
Serotonin metabolism in the ventromedial prefrontal cortex was increased and stress-related emotional behavior reduced in the forced swim test. Another study showed that administration of live M. vaccae reduced anxiety-like behaviors and improved performance in the Hebb–Williams complex maze. It was subsequently hypothesized that the antigens elicited an effect on the immune system, through which serotonin pathways were changed following ingestion of these bacteria (through the Th1 and Treg pathways), resulting in an anxiolytic response and improved performance in a land maze (Matthews and Jenks, 2013).
More recent research found that M. vaccae-pretreated mice responded to a larger, more aggressive animal with a more proactive coping strategy. Furthermore, M. vaccae elicited an anxiolytic response (Reber et al., 2016).
Although mouse models are very popular for in vivo immunological experimentation, there are significant differences between the two species in immune system development, activation, and response to a challenge, in the innate as well as the adaptive arms (Mestas and Hughes, 2004). It will, therefore, be prudent to establish whether a similar immunological and subsequent anxiolytic effect is present in humans treated with M. vaccae.
The microbiome and treatment response
The microbiome is not only an attractive therapeutic target in the treatment of psychiatric disorders but it could also be involved in treatment response and adverse drug reactions, which often occur in psychiatric patients. A preclinical study, using wild-type female C57BL/6J mice, investigated whether risperidone treatment (known to induce metabolic side effects) altered the gut microbiome profile and whether this shift was involved in the metabolic side effects of the drug (Bahr et al., 2015). As expected, the risperidone-treated mice exhibited significant weight gain, attributed to reduced energy expenditure, which was correlated with an altered gut microbiome. Fecal transplant, as well as transplantation of only the phage fraction, from risperidone-treated mice to naive recipients, resulted in a reduction in total resting metabolic rate and weight gain in the recipients, as a result of suppression of nonaerobic metabolism.
This study revealed the role of the gut microbiome in risperidone-induced weight gain, associated with altered nonaerobic resting metabolism (Bahr et al., 2015). In addition, it underscores the importance of taking treatment into account in case–control studies. Research efforts should be aimed at determining the role of the microbiome in the response to treatment in anxiety- and trauma-related disorders.
Clinical research frontiers
The effects of probiotics on the structure and function of the human gut microbiota have only been studied with a few specific bacterial strains; the effects of these treatments on clinical symptoms remain to be fully elucidated (Sanders et al., 2013). Tillisch et al. (2013) investigated the effects of probiotics on brain function in healthy female participants on a 4-week, chronic probiotic treatment (containing Bifidobacterium animalis subspecies Lactis, Streptococcus thermophiles, Lactobacillus bulgaricus, and Lactococcus lactis subspecies Lactis). Reduced brain response to the emotional faces attention task, particularly in sensory and interoceptive regions, was evident in participants who ingested the probiotic (4-week treatment). Probiotic ingestion was also associated with changes in midbrain connectivity, however, no differences in mood were observed between the treatment groups (Tillisch et al., 2013).
This study illustrated that treatment with this particular probiotic affected activity of brain regions that control central emotional and sensory processing.
In a randomized, double-blind, placebo-controlled trial, a Lactobacillus-containing probiotic, decreased anxiety but not depression symptoms in patients with chronic fatigue syndrome. Increased relative abundance of Bifidobacterium and Lactobacillus was also detected in stool samples of the treatment group (Rao et al., 2009). However, this study used culture techniques to determine the microbial composition of stool samples, thereby limiting the findings to only culturable microbes. Another study investigated the effects of a Lactobacillius- and Bifidobacterium-containing probiotic on mood and cognition in healthy individuals and found that the percentage decrease in the total Hospital Anxiety and Depression Scale score was greater in the probiotic-treated group, but that there was no difference in the subscale scores (Messaoudi et al., 2011).
A study by Diop et al. (2008) investigated the effects of a probiotic preparation (L. acidophilus and B. longum) on stress-induced symptoms in individuals affected by chronic stress. Abdominal pain and nausea/vomiting symptoms were significantly reduced in the probiotic group, however, physical and psychological symptoms were unaffected (Diop et al., 2008). A 2-week, controlled trial of Clostridium butyricum treatment resulted in significantly decreased Hamilton Anxiety Scale scores, as well as lower serum CRH levels before laryngeal cancer surgery compared to the placebo-treated sample (Yang et al., 2016).
A double-blind, placebo-controlled pilot study investigated the effects of an 8-week treatment of Lactobacillus casei strain Shirota (LcS) on psychological, physiological, and physical stress responses in medical students undertaking an authorized nationwide examination for promotion. One day before the examination, salivary cortisol and plasma L-tryptophan levels were significantly increased in the placebo group only, which was associated with a significant increase in anxiety. The probiotic group had significantly higher fecal serotonin levels compared to the placebo group, 2 weeks after the examination.
Moreover, the subjects receiving probiotics experienced significantly fewer physical symptoms compared to the placebo group during the pre-examination period and the intervention period. These results suggest that daily consumption of fermented milk containing LcS by healthy subjects during stressful time periods may decrease the onset of physical symptoms (Kato-Kataoka et al., 2016).
A recent study applied a double-blind design to investigate whether an 8-week-long treatment with a probiotic preparation, containing Lactobacillus helveticus and B. longum, had an effect on mood, stress, and anxiety in an antidepressant-naive sample of 79 individuals, selected for low mood (based on self-report data) (Romijn et al., 2017). The study found no evidence that the probiotic formulation had a positive effect on mood, or in moderating the levels of inflammatory and other biomarkers.
The authors hypothesized that the severity, chronicity, or treatment resistance of their sample may have contributed to the lack of effect on mood symptoms (Romijn et al., 2017). Another double-blinded, randomized, placebo-controlled clinical trial evaluated the effect of a 12-week treatment of Lactobacillus reuteri on digestive health and well-being in 290 older adults (>65 years). L. reuteri elicited no persistent significant effects on the primary or secondary outcomes of the study and the RCT failed to show a consistent improvement in digestive health, well-being, stress, or anxiety following a 12-week daily probiotic supplementation containing L. reuteri (Östlund-Lagerström et al., 2016).
These studies illustrate there is some potential for probiotics to influence CNS functioning and behavior. Furthermore, these results also underscore probiotic strain-specific effects. Probiotic trials require careful design as several factors may influence the outcome of such interventions, including confounding factors and matching of patients and controls. Comparing the results of these studies is complicated by the between-study differences, such as differences in probiotic strains, treatment duration, outcome measures, as well as gender and age distribution. In addition, samples sizes are relatively small and larger cohorts would be required to verify these findings. Well-designed trials in cohorts with anxiety- and trauma-related disorders will shed more light on the potential for pre- and probiotic treatments for the relief of symptoms in these patients.
Approaches to Studying the Functional Microbiome
While the above review aimed to examine the ways in which the gut microbiome might play a transformative role for mental health pathogenesis and mental health maintenance, we think that the following methodologies to study the microbiome are in order for the interested reader who wishes to take on this new line of research in biological psychiatry and integrative holistic medicine.
The majority of studies discussed thus far used 16S rRNA sequencing to understand the taxonomic distribution and diversity of enteric microbial communities in health and disease. However, to progress from mere phylotyping to functional network analyses, and to identify proteins and metabolites produced by the microbial communities, several meta-omics approaches can be utilized.
Metagenomics
Shotgun metagenomics involves sequencing the collection of genomes present in an ecosystem (Handelsman et al., 1998) and allows characterization not only of the taxonomic composition but also of the functional metabolic potential of the microbiota and reconstruction of microbial metabolic pathways. Although sequencing full genomes is more costly than 16S sequencing, metagenomics provides a wealth of information about the gut microbiota and its functions.
Metagenomic analyses in healthy individuals from large population-based studies paved the way for future studies in a clinical context. The MetaHIT and HMP projects revealed similarities in functional gene profiles among individuals despite significant variation in taxonomic composition (Human Microbiome Project Consortium, 2012; Qin et al., 2010). This indicates the presence of a functional core microbiome with conserved molecular activities, more than a taxonomic core microbiota, which would consist of a conserved group of phylotypes. An updated catalog of the genes in the human gut microbiome containing data from all major large-scale metagenomic projects has recently been released, containing ∼10 million genes (Li et al., 2014).
Zheng et al. (2016) transplanted the gut microbiota from major depression disorder (MDD) patients and healthy controls to germ-free mice, and after observing behavioral differences in the mice (including higher anxiety-like behavior in the open field test in mice receiving microbiota from MDD patients), performed metagenomic sequencing on cecum samples. Most of the discriminating genes were related to carbohydrate and amino acid metabolism; mice with depression microbiota had, for example, increased starch, sucrose, and glutamate metabolic potential but reduced tryptophan and tyrosine synthesis potential. This highlights the importance of examining the functional potential of the microbiota to obtain insights into how the gut microbiome influences disease.
Metatranscriptomics
In metatranscriptomics, the RNA transcript pool expressed by a microbial community is analyzed using RNAseq. The presence of a gene in a metagenome does not guarantee its expression, and therefore, metatranscriptomics complements metagenomic data by identifying the genes expressed by the microbial community. It provides information on the active microbial processes at a given time point and allows changes in microbial gene expression over time and in response to perturbations, such as antibiotic usage, to be monitored. The main limitation of metatranscriptomics is that, as the mRNA transcript pool changes rapidly, it is uncertain how well the recovered RNA from stools represents the processes that were active in the ileum and colon, and not due to sampling-induced stress conditions. An interesting approach is dual RNAseq, where the host and microbiota transcriptomes are analyzed together.
The application of metatranscriptomics to study microbiota gene expression in health and disease is still rather limited. The importance of taking the metatranscriptome into account when performing microbiota studies was illustrated in a study of the human gut microbiota in 10 healthy individuals. They showed that transcripts for carbohydrate metabolism, energy production, and synthesis of cellular components were overrepresented compared to what would be expected based on their gene copy number in the metagenome, while activities such as lipid transport metabolism were underrepresented in the metatranscriptome compared to the metagenome (Gosalbes et al., 2011).
Metaproteomics
Metaproteomics is a high-throughput approach to identify the entire protein pool within complex, microbial habitats. It provides information on the metabolic processes that are active, and reveals how they are affected by perturbations, such as inflammation or disease conditions. Metaproteomics, therefore, provides a more direct insight into the functional composition of the microbiota compared to metatranscriptomics, as mRNAs are subject to posttranscriptional modification. In addition, the metaproteome is more stable than the metatranscriptome and thus less prone to sampling-induced alterations.
Metaproteomics involves cellular lysis and enzymatic digestion of all accessible proteins in a particular sample to produce peptide fragments that are separated by liquid chromatography and subjected to tandem mass spectrometry (LC-MS/MS). The mass and spectra of the peptides are subsequently quantified and compared to reference protein databases (predicted from genomic sequence information). Although metaproteomics is a powerful tool to characterize the function of complex microbial communities, some factors need to be taken into account, such as host-specific biases and choice of sequence databases for protein identification [the reader is referred to a review by Tanca et al. (2016) regarding investigation of variables concerning database construction and annotation and how it impacts metaproteomic results].
Metaproteomic analyses from fecal samples retrieve an important proportion of human proteins, but filtering strategies have been put forward to fractionate microbial cells from human cells and enhance microbial protein identification (Xiong et al., 2015). Tanca et al. (2016) provide guidelines on how to design gut microbiota studies to perform metaproteomic data analysis. They encourage the use of multiple databases and annotation tools.
Metaproteomic investigations of gut microbial communities are currently relatively limited; only a few studies have investigated the metaproteome in humans, including investigations in a healthy adult, monozygotic twin pair (Verberkmoes et al., 2009), a longitudinal investigation in healthy female participants (Kolmeder et al., 2012), Crohn's disease patients (Erickson et al., 2012; Juste et al., 2014), obese individuals (Ferrer et al., 2013), the infant gut (Xiong et al., 2015), and the preterm infant gut (Brooks et al., 2015; Young et al., 2015).
By using clusters of orthologous groups (COGs) to catalog identified proteins (Tatusov et al., 2000), Verberkmoes et al. (2009) found an uneven distribution of relative abundances of each COG in the metaproteome relative to metagenome. The metaproteome was enriched in proteins involved in translation, energy production, and carbohydrate metabolism, while proteins involved in cell division, inorganic ion metabolism, cell wall and membrane biogenesis, and secondary metabolite biosynthesis were less present than in the metagenome. Similar to what was observed using metatranscriptomics (Gosalbes et al., 2011), the findings highlight the fact that in situ functional activities (as measured by metaproteomics) can be distinct from predictions from metagenome information alone (Kolmeder et al., 2012; Verberkmoes et al., 2009).
Metabolomics
Metabolomics involves the metabolic profiling of biological fluids, such as serum, urine, or fecal water, using spectroscopic techniques to enable either global metabolite analysis (untargeted approach) or the measure of a selected metabolite (targeted approach). MS-based techniques, often preceded by separation techniques such as gas chromatography or high-performance/ultra-performance liquid chromatography, facilitate the discrimination of metabolites based on their mass to charge (m/z) ratio. Existing databases of m/z values, such as METLIN, are interrogated to identify metabolites (Smith et al., 2005). Another available method that is particularly popular for high-throughput studies is that of 1H nuclear magnetic resonance (1H NMR) spectroscopy. 1H NMR uses chemical shift (i.e., the resonant frequencies of atomic nuclei relative to a reference standard) after perturbation with radiofrequency pulses to identify chemical structures (Holmes et al., 2011).
The potential of metabolomics was illustrated in a gnotobiotic mouse study that colonized mice with a 15-species model human gut microbiota. Following introduction of a fermented milk product (containing five sequenced bacterial strains), no significant changes in the metagenome were observed, however, metatranscriptomics of fecal samples and MS of urinary metabolites indicated the effects of the milk product on the expression of microbial enzymes involved in carbohydrate metabolism (McNulty et al., 2011).
In light of the observed discrepancies between effects on microbial community structure relative to effects on gene expression and metabolism, the need for complementary approaches beyond 16S-based phylotyping is emphasized. Although bioinformatically challenging, multivariate computational modeling allows for the integration of metagenomic, metatranscriptomic, and metaproteomic/metabolomic profiles to provide insight into microbial functionality. The application of such approaches is quite challenging and would require the concerted efforts of experienced bioinformatic specialists, biostatisticians, mathematicians, clinicians, and molecular biologists to unravel the role of the functional microbiome in disease. In future, these promising strategies can be used to obtain a holistic overview of the role of the functional microbiome in anxiety- and trauma-related disorders.
Interactors of the Gut Microbiome
Other factors that shape the gut microbiome should also be considered when interpreting microbiome data, especially in the context of complex disorders. These include, but are not limited to, host factors, including host genome (Davenport, 2016), host epigenome (Liu et al., 2016a), external factors such as environmental microbiome, and constituents of the gut microbiota, such as the gut virome (Ogilvie and Jones, 2015) and parasitic gut infections (Molloy et al., 2013) (Fig. 1).
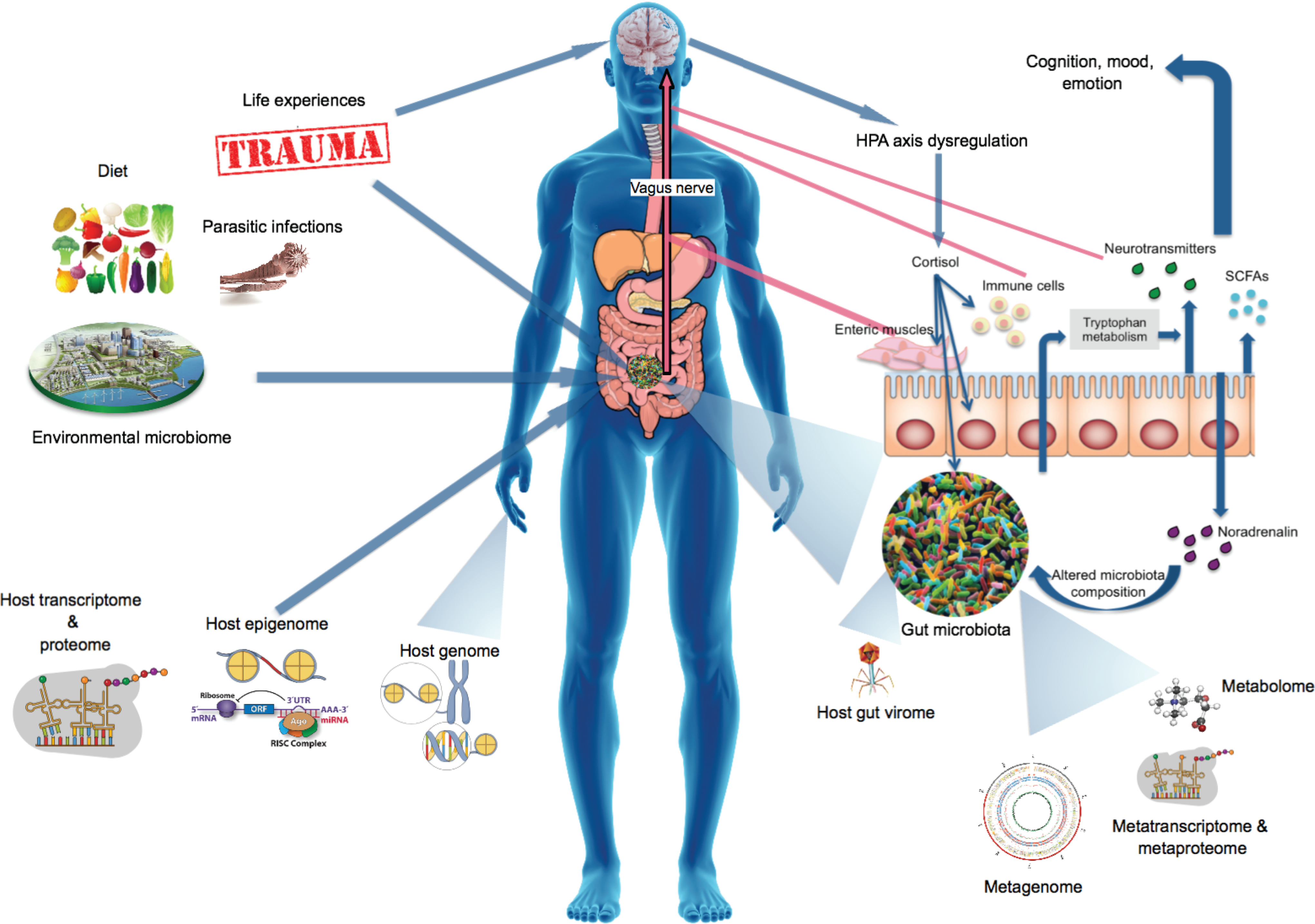
The human interactome encompasses the gut microbiome; its genes, proteins, and metabolites; and host factors and external environmental factors that concomitantly shape the microbiome and influence health and disease. The MGB axis contains pathways through which the microbiota influences the CNS, cognition, and mood. Furthermore, microbially produced proteins and metabolites can influence the host stress response system, CNS functioning, and the host epigenome and transcriptome. Traumatic experiences and stress can also alter the gut microbiota via HPA axis dysregulation and subsequent release of stress hormones or neurotransmitters that influence gut physiology, microbiota habitat, and composition and bacterial gene expression. CNS, central nervous system; HPA, hypothalamic–pituitary–adrenal; MGB, microbiota–gut–brain.
The host genome
Results from murine models showed that host genotypes play a role in shaping microbiota composition (Bongers et al., 2014; Campbell et al., 2012; Hildebrand et al., 2013). Turnbaugh et al. (2009) and Yatsunenko et al. (2012) investigated whether this was also the case in humans. They investigated the heritability of the gut microbiome using monozygotic and dizygotic twins, while controlling for environmental factors. Both studies concluded that there were no statistically significant differences between monozygotic and dizygotic twins, however, both studies were underpowered (small sample sizes). Larger follow-up studies of these twin data sets revealed that host genetic variation did have an influence on microbial composition. They found Christensenellaceae to be the most heritable taxon, while Bacteroidetes was more susceptible to environmental influences (Goodrich et al., 2014, 2016). They also showed that highly heritable taxa were associated with higher levels of temporal stability, emphasizing the importance of these taxa to the host (Goodrich et al., 2016).
Folseraas et al. (2012) examined the role of host genetic loci associated with primary sclerosing cholangitis (PSC) and the effect on the biliary microbial community composition in PSC patients. They showed that secretor status and genotype of the Fucosyltransferase 2 (FUT2) gene (rs601338) significantly influenced biliary microbial community composition, as it was associated with a significant increase in the abundance of Firmicutes and significant decrease of Proteobacteria. Furthermore, decreased alpha diversity was noted in the heterozygous state compared to both homozygous genotypes. Knights et al. (2014) showed that host genetics influence the microbiome in inflammatory bowel disease (IBD) when they detected a significant association between nucleotide binding oligomerization domain containing 2 (NOD2) risk allele count and increased relative abundance of Enterobacteriaceae.
This finding was confirmed in two additional cohorts. These two studies emphasized the impact of the genotypes of these two genes and their associated bacterial taxa alterations as risk factors for PSC and IBD. These results suggest complex interactions between host genetics, subsequent altered functional pathways, and the composition of the microbiome. These studies were able to identify genome–microbiome associations in diseased cohorts using a candidate gene approach; however, recent studies have identified genome-wide, statistically significant genetic loci that influence gut microbiota composition (Blekhman et al., 2015; Bonder et al., 2016; Davenport, 2016; Goodrich et al., 2016; Wang et al., 2016).
Several of these studies found that specific bacterial taxa, such as Bifidobacterium, are inheritable and correlate with specific host genotypes (Blekhman et al., 2015; Davenport, 2016; Goodrich et al., 2016). Goodrich et al. (2016) linked the lactase (LCT) gene locus, which encodes the enzyme lactase that hydrolyzes lactose, to Bifidobacteria, which metabolizes lactose in the gut. They discovered that lactase “nonpersisters” (inactive lactase enzyme) harbored lower levels of Bifidobacterium compared to lactase “persisters” (active lactase enzyme) possibly due to higher lactose levels in the gut of lactase nonpersisters.
Similarly, Bonder et al. (2016) found an association between a functional LCT SNP and the Bifidobacterium genus. They also found nine genetic loci associated with microbial taxonomies and 33 loci with microbial pathways, and reported on associations between bacterial taxa and metabolic loci, suggesting that the gut microbiota could be a mediating factor in the link between host genetics and immunological and metabolic phenotypes (Bonder et al., 2016). Blekhman et al. (2015) and Davenport (2016) found that host genetic variation in immunity-related pathways, such as IL-2, is correlated with microbiome composition, and host genes and variants that are correlated with microbiome composition are enriched in genes associated with complex diseases that have been linked to the microbiome (such as irritable bowel syndrome (IBS) and obesity-related diseases).
A recent study found genome-wide significant associations between gut microbial characteristics and several host genetic factors, including the vitamin D receptor (VDR) gene. They calculated that, in total, the host genetic loci contributed to 10.43% of β diversity and nongenetic factors (including age, sex, BMI, smoking status, and dietary patterns) explained 8.87% of the variation in the gut microbiome. They were also able to replicate genetic associations reported in previous studies (such as FUT2, NOD2, and LCT), but found that they contributed less to the overall microbial variation (Wang et al., 2016).
Future studies could investigate combined host genome–microbiome data in anxiety- and trauma-related disorders, using candidate gene and, given sufficient sample numbers, genome-wide association study (GWAS) approaches to shed more light on this complex link between host genome and microbiome.
The host epigenome
Epigenetics literally translates to “outside conventional genetics” and it investigates the stable alterations in gene expression not attributable to changes in DNA sequence (Bjornsson et al., 2004). Epigenetic processes include DNA methylation, posttranslational modification of histone proteins, genomic imprinting, and noncoding RNAs (including microRNA [miRNA], small interfering RNA, and long noncoding RNA).
miRNAs are a class of small, noncoding RNAs that epigenetically modulate gene expression. A recent study discovered that host fecal miRNAs, produced by the gut epithelial and Hopx+ cells, regulate bacterial gene expression and growth (Liu et al., 2016a). miRNAs are usually synthesized in the nucleus and processed in the cytoplasm where they perform their function.
However, there is evidence that miRNAs exist in extracellular compartments and circulate in body fluids (Weber et al., 2010). Abundant levels of miRNAs have been detected in mouse and human fecal samples (Ahmed et al., 2009; Link et al., 2012; Liu et al., 2016a). Liu et al. (2016a) showed that extracellular fecal miRNAs are mainly produced by the IEC and Hopx+ cells and that fecal miRNAs can enter bacteria and regulate bacterial gene transcripts and directly affect gut bacterial growth. In addition, they illustrated that deficiency of IEC miRNAs increases the dissimilarity of the gut microbiota and alters intestinal barrier integrity. Transplanting wild-type fecal miRNAs in IEC miRNA-deficient mice restored the fecal microbes and rescued dextran sulfate sodium-induced colitis in a colitis animal model.
The authors explained that this miRNA-mediated bacterial regulation was different from traditional miRNA regulation in eukaryotic cells that mainly result in posttranscriptional repression (including mRNA cleavage, destabilization, and a reduced translation efficiency) (Bartel, 2009; Fabian et al., 2010). In this case, the regulation of bacterial targets by host miRNAs extended to rRNA (16S rRNA) and ribozyme (RNaseP), and the effect included a decrease, as well as enhancement of the transcripts. The mechanism through which miRNAs regulate gene expression and affect bacterial growth probably depends on the function of the target gene (Liu et al., 2016a).
Their results highlight the role of host fecal miRNAs in targeting and regulating the gut microbiota and the possibility of using miRNAs as a tool to manipulate the microbiome to benefit the host. Such investigations can easily be performed in human stool samples to determine how fecal miRNAs regulate bacterial gene expression and growth, and should be performed to determine whether particular miRNA profiles can be associated with a dysregulated microbial composition in the context of psychiatric disorders.
The microbiome also has the ability to alter certain host epigenetic processes (Paul et al., 2015). Bacteria can produce epigenetically active metabolites such as folate, butyrate, and acetate, and therefore have the ability to influence host DNA methylation patterns. For instance, folate (produced by Bifidobacterium spp., among other bacterial genera) is a methyl donor and is crucial for the production of S-adenosylmethionine, which in turn is a methyl-donating substrate for DNA methyltransferases (Hesson, 2013).
Histone acetylation entails the transfer of an acetyl group from acetyl coenzyme A (acetyl-CoA) to lysine residues, with the subsequent production of CoA (Roth et al., 2001). The histone acetylation process is regulated by the tricarboxylic acid (TCA) cycle and acetylation is primarily associated with transcriptional activation, due to increased accessibility of nucleosomal DNA to transcription factors. Histone deacetylases (HDACs) remove acetyl groups from lysine residues. The gut microbiome produces short-chain fatty acids that are used in the production of ATP via the TCA cycle. In addition, some of the metabolites produced by the gut microbiome, such as butyrate and propionate, can inhibit HDACs, thereby influencing the histone acetylation process and ultimately transcription (Paul et al., 2015).
Methods such as DNA methylation arrays, bisulfite sequencing (Kurdyukov and Bullock, 2016), and chromatin immunoprecipitation (ChIP) microarrays (ChIP-chip) (Ren et al., 2000) map DNA methylation, functional status of DNA-binding proteins, histone modifications (Robyr and Grunstein, 2003), and nucleosome distribution (Ozsolak et al., 2007) on a global scale. These techniques are routinely used and can be incorporated into microbiome studies to investigate whether the abundance of certain metabolite-producing bacteria is associated with, for instance, altered DNA methylation or histone profiles, which, in turn, could influence host gene expression profiles.
Despite these indications, it is evident that more research is warranted into the intricate relationship between the host epigenome and gut microbiome, how they influence each other, and their impact on the evolution and course of disease (Fig. 1).
Environmental microbiome
Although consideration of a role for the environmental microbiome, either the microbiome of outdoor environments or the microbiome of the built environment (MoBE), is in its infancy, there is growing evidence that the environmental microbiome may play a role in determining mental health outcomes. Although outside the scope of this review, this emerging field has been extensively reviewed (Hoisington et al., 2015; Lowry et al., 2016; Stamper et al., 2016) and can be included in future microbiome-interactome models to better understand health and disease (Fig. 1).
Constituents of the gut microbiota
The gut virome
The gut virome consists of viruses, or virus-like particles, that coexist with bacteria in the gut. Viruses are at least 10 times more abundant in the human body than the microbes that form part of the bacterial microbiome (Mokili et al., 2012). The virome includes viruses that infect host cells or other organisms (such as bacteriophages and plant viruses) as well as virus-derived elements in our chromosomes. Bacteriophages are prokaryotic viruses that infect bacteria and alter their metabolism and replication (Breitbart et al., 2003, 2008; Minot et al., 2011; Reyes et al., 2010). Bacteriophages have the ability to facilitate gene transfer between strains and species (transduction) and thereby influence community function.
Phages can also confer some of their crucial functional attributes to their bacterial hosts, such as the production of virulence factors and toxins as well as genes that provide metabolic flexibility (Brüssow et al., 2004; Fuhrman, 1999; Suttle, 2007; Wommack and Colwell, 2000). The phage–host relationship is a dynamic coevolutionary interaction, which forms an integral part in the evolution of the bacterial hosts (Paterson et al., 2010). Phages are therefore considered as a strong driving force of ecological function and evolutionary change in prokaryotes (Koskella and Brockhurst, 2014). Although a comprehensive discussion of the phage–host relationship is beyond the scope of this article, the reader is referred to an insightful review by Ogilvie and Jones (2015).
Yolken et al. (2014) detected DNA homologous to the chlorovirus, Acanthocystis turfacea chlorella virus 1 (ATCV-1), in 43.5% of the oropharyngeal samples of their healthy cohort. Chloroviruses infect certain eukaryotic green algae but have never been shown to infect humans or to be part of the human virome. Individuals that harbored the ATCV-1 DNA showed a significant decrease in the performance on cognitive assessments of visual processing and visual motor speed.
Further investigations in a mouse model showed that inoculation of mice with ATCV-1 DNA resulted in decreased performance in several cognitive domains. Exposure to ATCV-1 DNA also induced altered gene expression profiles in the hippocampus in pathways related to synaptic plasticity, learning, memory, and immune response to viral exposure. These results implicate immune response as a possible mechanism underlying the cognitive deficits to ATCV −1.
The authors hypothesized that immune activation resulted in proinflammatory cytokine secretion, subsequently affecting neuronal functioning, which in turn resulted in behavioral abnormalities. Shared and unique profiles of cytokine upregulation have been shown for various microbial infections (e.g., Borna virus vs. Toxoplasma), and therefore, unique signatures of cytokine expression might help to explain differential neurobehavioral outcomes of different microbial infections (Stewart et al., 2015).
Investigations into the role of the virome in psychiatric disorders are very limited. One study compared the bacteriophage genomes in the oral pharynx of individuals with schizophrenia to those of control individuals (Yolken et al., 2015) and found that Lactobacillus phage phiadh was significantly enriched in individuals with schizophrenia compared to controls. Phage phiadh was also associated with an increased prevalence of comorbid immunological disorders in individuals with schizophrenia.
In addition, individuals taking valproate had no detectable levels of Lactobacillus phage phiadh. Interestingly, valproate was previously shown to alter the gut microbiome and to change levels of microbial metabolites in an animal model of autism (de Theije et al., 2014). The mechanisms through which Lactobacillus phage phiadh is associated with schizophrenia and comorbid immunological conditions are not clear; however, the authors speculated that Lactobacillus phage phiadh probably modifies the level of its host bacteria, Lactobacillus gasseri, with subsequent effects on the host immune systems. Lactobacillus gasseri modulates the immune system by modifying the function of enterocytes, dendritic cells, and components of innate immunity (Luongo et al., 2013; Selle and Klaenhammer, 2013).
The authors concluded that the therapeutic altering of bacteriophages could provide new means of treating schizophrenia and some of its comorbid immunological diseases (Yolken et al., 2015).
The relationship between changes in bacterial and viral communities is a novel area of investigation. The microbiome and the virome are affected by similar environmental stimuli, evidenced by covariation of the virome with the bacterial microbiome in response to diet (Minot et al., 2011). The virome also plays a crucial role in the regulation of intestinal immunity and homeostasis (Norman et al., 2015). The virome can induce continuous, low-level immune responses without triggering any apparent symptoms. It is therefore plausible that variations within the systemic and local gut virome could influence the host gut microbiome as well as host immunophenotype (Virgin, 2014) and ultimately affect CNS functioning (Fig. 1).
Parasitic infections
The mammalian gut is not only populated by microscopic members of the microbiota but could also include larger organisms, such as parasitic nematodes or worms. A study of a murine model infected with T. muris showed that these parasites compete for nutrients in the intestines of infected animals and can directly interact with bacterial members of the microbiota during the parasitic life cycle to promote hatching of parasite eggs. These parasites also influence immune functioning through factors such as excretory–secretory products, which modulate cytokine production, immune-cell recruitment, basophil degranulation, and interfere with toll-like receptor signaling (Hayes et al., 2010).
Interestingly, one of the risk factors for the development of schizophrenia and a contributor to dysbiosis and altered immune reactivity is infection with the parasite Toxoplasma gondii (Molloy et al., 2013; Torrey et al., 2007). Causal mechanisms linking infection with disease risk are speculative, but could, in part, be attributed to the tachyzoites forming cysts in the brain and the establishment of a chronic infection (Carruthers and Suzuki, 2007). Two meta-analyses found elevated levels of T. gondii antibodies in patients with schizophrenia compared to healthy controls (Torrey et al., 2007, 2012). Infection by T. gondii results in gastrointestinal inflammation, which subsequently triggers an innate immune reaction, including activation of complement C1q; C1q in turn plays a role in synaptic pruning (Chu et al., 2010).
Since pathogen proteins closely resemble those of humans, the immune attack directed toward the pathogen may also result in the development of pathogen-derived autoantibodies. In addition, this inflammation influences endothelial barrier permeability and could therefore facilitate translocation of gut bacteria into systemic circulation, resulting in further dysbiosis and immune reactivity. T. gondii also induces major perturbations on gut microbiota composition (Molloy et al., 2013). These findings suggest that individuals living in areas with higher exposure to such parasites could have significantly different structural and functional configurations of gut-associated immune systems compared to individuals without these exposures. Such effects should be taken into account when interpreting microbiome findings from populations with higher exposure, especially in psychiatric patients.
Future Perspectives
There have been major advances in the field of microbiome research, including large-scale population-based studies, such as the Human Microbiome Project, MetaHIT, Lifelines-DEEP, and the Flemish Gut Flora Project, which have identified factors that are linked to gut microbiota composition. Studies have reported that ethnicity and lifestyle could influence gut microbial profiles (Chong et al., 2015; Liu et al., 2016b), and therefore, future studies should include more population-based studies in ethnically diverse groups to clarify this association and to determine the composition of a healthy gut microbiome in a particular population.
Furthermore, certain pitfalls should be taken into account when performing microbiome analyses. These include experimental design, such as selection of 16S rRNA target region and sequencing platform (Tremblay et al., 2015), sample collection, storage (Vogtmann et al., 2017) and extraction methods, inclusion of positive and negative controls (Weiss et al., 2014), taking cage effects into consideration in animal models (Hildebrand et al., 2013), and the use of discovery and validation cohorts (Forslund et al., 2015; Sabino et al., 2016) as well as robust data analyses that incorporate power calculations (Kelly et al., 2015), appropriate reference genome databases (Balvočiūtė and Huson, 2017; Forster et al., 2016), correction for multiple comparisons (Benjamini and Hochberg, 1995), and confounders (Falony et al., 2016).
Meta-omic technologies (such as metatranscriptomics, metaproteomics, and metabolomics, as discussed earlier) are increasingly used in the laboratory and enable us to interrogate the taxonomic and functional composition of the microbiome, as well as protein and metabolite synthesis to determine their role in health and disease.
However, these techniques are not without challenges, which mirror those discussed for microbiome analyses (including sample collection, storage, processing, data analysis strategies, and use of appropriate databases and analysis pipelines). In addition, there is a need for meta-information (i.e., databases of information on sample origin, collection and storage, and experimental and analytical conditions) (Weckwerth and Morgenthal, 2005) and the integration of multiple data sets arising from multi-omic outputs (Abram, 2015), for example, by using network-based approaches, such as the 48-h multi-omic pipeline developed by Quinn et al. (2016) [also refer to review by Aguiar-Pulido et al. (2016)].
Recent research has also indicated a role of the host in shaping the gut microbiome content, including host genetic variation and host epigenetic factors, as well as the host virome. Furthermore, the effect of exposure to environmental microbes and parasitic gut infections also plays a role in modulating the immune system as well as the gut microbiome. The feasibility of host-genome-epigenome-microbiome investigations lie within our grasp, as evident in the literature presented in this review; however, the combined investigation in a single cohort is yet to be endeavored.
Challenges include the high cost of multi-omic investigations in large, well-characterized cohorts as well the need for sophisticated bioinformatic pipelines and mathematical models to integrate output from several omic data sets. Moreover, in an attempt to attain a whole systems overview (including the functional microbiome as well as host and environmental factors that influence and interact with the microbiome), we require mathematical models and statistical approaches to enable the meaningful biological interpretation of multi-omic outputs.
Animal models provide strong evidence for the role of the gut microbiome in regulating anxiety- and stress-related phenotypes, and the potential to target the gut microbiome to alleviate anxiety. However, few human studies of the microbiome in anxiety- and stress-related disorders have been published. As effective probiotic treatments in animal models have not translated well to humans, gut microbiome studies of human subjects with anxiety disorders are warranted, before we can even attempt to understand the complexities of the functions, genes, pathways, proteins, and metabolites of the gut microbiome.
Furthermore, to show causation and to understand the mechanisms through which dysbiosis influences disease, longitudinal studies are needed, preferably with birth cohorts or pre- and postdeployment cohorts, to track disease progression before onset. Such study designs would also enable the investigation of the role of the gut microbiome in treatment response.
Probiotic intervention studies in humans suggest that the gut microbiome could be targeted to alleviate anxiety- and stress- or trauma-related outcomes. However, interpretation of these findings is impeded by several limitations, including small sample sizes and confounders such as different populations/ethnicities, gender bias, different probiotic strains (or combinations of strains) at different doses and for different treatment durations, and differences in outcome measurements. More concerted efforts to recruit large cohorts and adopt standardized approaches could yield more insights into how the gut microbiome can be targeted to alleviate anxiety- and stress- or trauma-related outcomes.
Future studies should also address aspects such as the host's baseline microbiota composition and whether it predicts response to the probiotic treatment, possible effects of the probiotic vehicle, dose/response effects, and the stability of the treatment response. These studies should preferably use a longitudinal design, even beyond the standard treatment duration in clinical trials, to fully assess the long-term effects of microbial manipulation on behavior. Once we understand how the microbial composition is associated with disease, the aforementioned recommendations can be used to design more targeted microbial therapies in the near future. Once successful therapies have been designed and proven to be useful, future investigations could also utilize imaging data, such as fMRI and spectroscopy, to measure functional brain changes pre- and post-prebiotic, synbiotic, probiotic, or antibiotic interventions.
Another complicating factor in the investigation and understanding of anxiety- and trauma-related disorders is phenotypic heterogeneity and the high prevalence of psychiatric and medical comorbid disorders, including depression (Elhai et al., 2008; Rytwinski et al., 2013) and metabolic diseases (Kahl et al., 2015; Meurs et al., 2016). Anxiety- and trauma-related disorders, and their common comorbidities, have been associated with increased inflammation (Zass et al., 2017), suggesting that the gut microbiome could play a role in comorbidity.
Careful study designs, using large cohorts and inclusion of appropriate controls, will be required to understand the underpinnings of comorbidity. Furthermore, since a plethora of environmental variables has been shown to alter the microbiome, the collection of metadata should be extensive and thorough and variables should be tested for association with microbial composition to correct for the effects of confounding variables during data analysis.
This review focused on the gut microbiome in the context of the human interactome. However, it should be noted that other microbial habitats include the mouth, skin, urogenital tract, and vagina, and that these could also play an intricate role in the human interactome. As an example, afferent signaling of bronchopulmonary immune activation to the CNS has been described (Hale et al., 2012; Lowry et al., 2016). Immune signals in the bronchopulmonary system reach the brain via the vagus nerve and sympathetic nerves, much like the afferents from the gastrointestinal system, but the specific targets of the bronchopulmonary afferents in the brain are distinct from the specific targets of the gastrointestinal afferents in the brain (Hale et al., 2012).
Thus, peripheral signals arising from the microbiome in the bronchopulmonary system are not redundant with those arising from the gastrointestinal system, and may have unique cognitive and affective functions. Similar arguments could be made for the skin (Belkaid and Segre, 2014) and oral microbiomes (Castro-Nallar et al., 2015). This review, however, focussed on the gut microbiome due to its established involvement in the MGB and its implications for anxiety- and stress-related disorders. Future studies could further investigate the role of the microbiome in other body sites in the context of anxiety- and stress-related disorders.
Great efforts are being made to discover the missing heritability in complex disorders, such as anxiety- and trauma-related disorders. Investigations of gene–gene and gene–environment interactions (Nugent et al., 2011), copy number variations (Bersani et al., 2016; Fung et al., 2010; Kawamura et al., 2011), and epigenetic factors (Cappi et al., 2016; Kim et al., 2017) have yielded some additional insights into the molecular etiology of these disorders. However, renewed interest and large-scale focus on microbial communities, investigation of the human interactome, which includes the (gut) microbiome composition, its genes, proteins, and metabolites, as well as host and environmental factors that shape the microbiome, have the potential to unravel the etiology of complex disorders and direct novel treatment strategies.
Footnotes
Acknowledgments
This work is based on research supported by the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation and the South African Medical Research Council.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.