
Abstract
Abstract
Molecular pathogenesis of oral cancers continues to be researched by omics systems science biotechnologies. Oral cancers rank as the 13th most common cancer globally. Notably, the burden of oral cancers from the Asian continent is 56.21%, with 26% of the burden contributed by India. Despite easy accessibility of the oral cavity and hence early detection of oral cancers, majority are diagnosed in advanced stages in the Asian countries. Innovation in oral cancer diagnostics, as well as theranostics for precision medicine, would aid their early diagnosis, prognosis, and treatment, not to mention discovery of novel molecular targets for drug development. This expert review offers an analysis of oral cancer biomarkers, including somatic mutations, deregulated expression, epigenetic regulation, and genomic variants associated with oral cancer. We also discuss the implications of the current and emerging oral cancer biomarkers with a view to clinical practice, global health, and make suggestions for the ways forward.
Introduction
C
Tobacco, areca nut, alcohol, and high risk human papillomavirus (HPV) 16/18 are major risk factors of oral cancers, with tobacco smoking and alcohol consumption as major risk factors in the western countries and tobacco chewing compounded with smoking and alcohol consumption highly prevalent in Asia (D'Costa et al., 1998; Rao et al., 2013; Saranath, 2000; Warnakulasuriya, 2009). The metabolites of tobacco include tobacco specific N′nitrosonornicotine (NNN) and 4-(methylnitrosoamino)-1-(3-pyridyl)-1 butanone (NNK), and areca nut alkaloids—arecoline and arecaidine, polycyclic aromatic hydrocarbons, volatile aldehydes and hydroquinones, and alcohol metabolite—acetaldehyde act as potent carcinogens (Hecht, 2003; Ribeiro et al., 2016). Generally, HPV positive oral cancers show a more favorable prognosis than the HPV-negative counterparts, perhaps due to an improved immune response toward the virus. However, correlation of viral pathology, molecular pathology, and histopathology in oral cancers is not clear (O'Rorke et al., 2012).
Leukoplakia, erythroplakia, and oral submucous fibrosis are the common precancerous lesions observed in several Asian countries, and 3–8.1% of the precancerous oral lesions transform to oral cancer (Scheifele and Reichart, 2003). The malignant transformation rate is 0.64% to 1.41% dependent on the site of oral cancer and 0.13% to 17.5% dependent on the premalignant lesion and grade of dysplasia (Amagasa et al., 2011; Dost et al., 2014). A dose–response association with the quantity, frequency, and duration of tobacco consumed has been associated with oral cancer development (Rao et al., 2013).
Innovation in oral cancer diagnostics, as well as theranostics for precision medicine, would aid their early diagnosis, prognosis, and treatment, not to mention discovery of novel molecular targets for drug development. This expert review offers an analysis of oral cancer biomarkers, including somatic mutations, deregulated expression, epigenetic regulation, and genomic variants associated with oral cancers. We also discuss the implications of the current and emerging oral cancer biomarkers with a view to clinical practice and global health and make suggestions for the ways forward.
A Global Public Health Perspective on Oral Cancers
In the last three decades, significant improvement in prognosis, survival, and mortality in oral cancers has not been observed, despite advances in treatment protocols of surgery, radiotherapy, chemotherapy, and biological therapy (Ribeiro et al., 2016). Primary prevention in oral cancer through health education and implementation of tobacco/areca nut/alcohol cessation programs at individual and community levels and aggressive implementation of chemoprevention in high incidence regions with persistent precancerous lesions may result in downgrading oral cancers with better prognosis. Secondary prevention, including screening of individuals with persistent precancerous lesions and early-stage oral cancer, may help reduce mortality and improve quality of life in oral cancer patients (Notani, 2000). The main obstacle to reduce morbidity and mortality of oral cancers is to change the lifestyle habits of individuals using tobacco, areca nut, and alcohol, a rather difficult proposition (Rethman et al., 2010).
Advances in biotechnology and bioinformatics have facilitated understanding of oral cancer biology, development of molecular biomarkers for screening high risk oral lesions, recurrence, and response to therapy. Besides, the molecular markers may enable identification of novel drug targets to inhibit specific molecules associated with oral cancer pathology, enabling effective intervention.
It is interesting to note that only a small proportion of individuals with high risk habits develop oral cancer as mentioned earlier (Scheifele and Reichart, 2003), similar to tobacco smokers and lung cancer; alcohol consumers, HBV/HCV infections, and liver cancer; and Helicobacter pylori and stomach cancer (Danaei et al., 2005). Tobacco smoking and alcohol cause more than 80% of oral cancers (Warnakulasuriya, 2009), whereas tobacco chewing and areca nut cause oral cancers with the attributable risk of 81% in Indian males (Notani, 2000). HPV positivity is increasing in oral cancers, particularly in the western population to up to 24.2% (Ndiaye et al., 2014). Whereas in oropharyngeal cancers, HPV positivity ranges from 56% in North America, 52% in Japan, 45% in Australia, 39% in northern and western Europe, and 13% in the rest of the world (Marur and Forastiere, 2016).
Oral cancer is a complex multifactorial disease with several molecular lesions driving the pathogenesis. The molecular lesions include somatic mutations, consequent deregulated expression, epigenetic modifications silencing the genes, and genomic variants influencing genes in the cellular pathways of malignant transformation as outlined in Figure 1. Tumor development and progression is a result of cumulative lesions in various pathways leading to DNA damage, excessive cell proliferation, inhibition of apoptosis, angiogenesis and invasion into the surrounding tissues, and distant metastasis (Fig. 1). The multistage pathogenesis model with parallel histopathological and molecular pathogenesis resulting in conversion of benign to premalignant lesions to oral cancer is indicated in Figure 2, adding to the concept proposed by Califano et al. (1996).
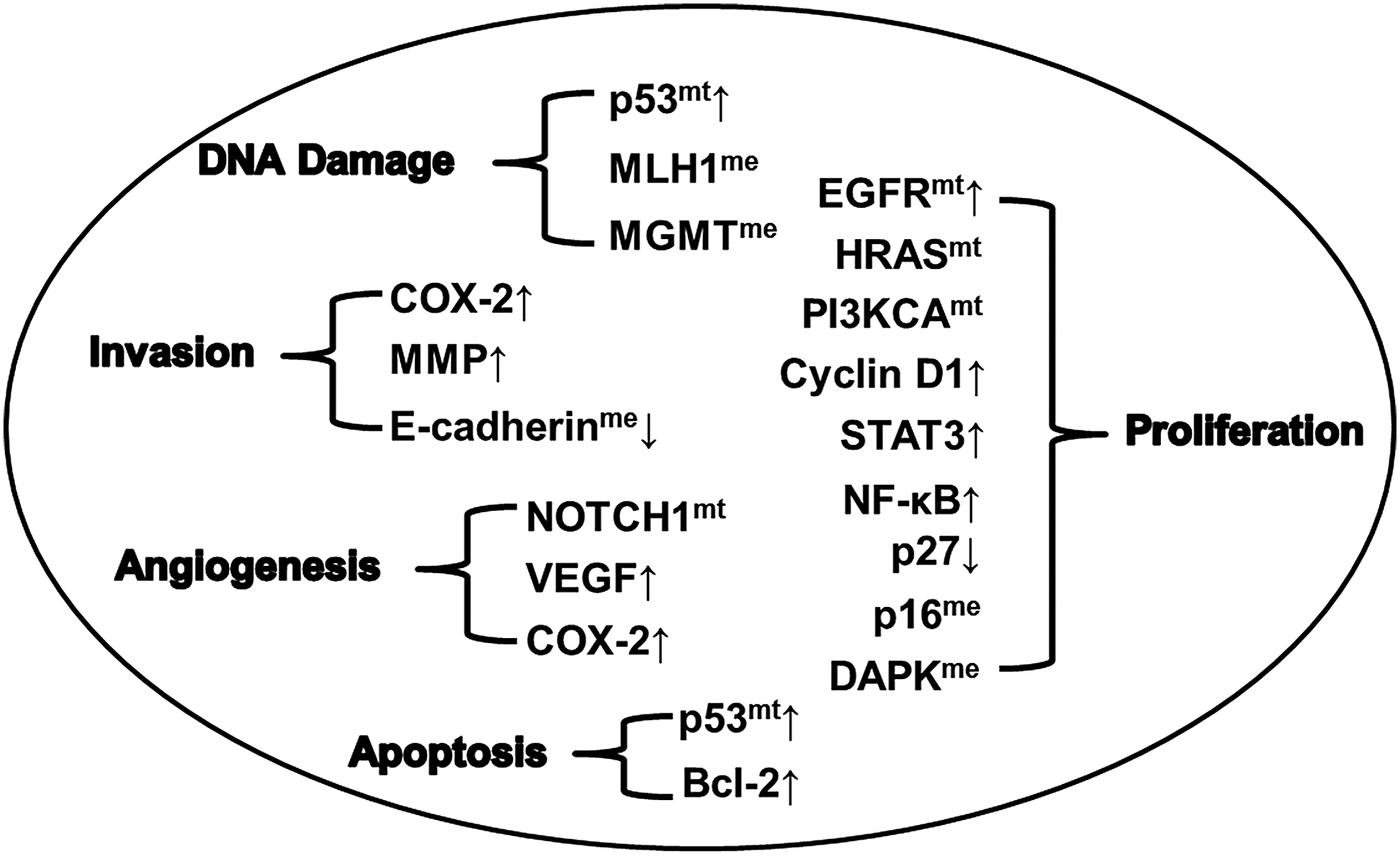
Molecular aberrations in critical genes associated with oral cancer pathogenesis. Tumor development and progression is a result of multiple molecular lesions in critical processes. The molecular lesions in multiple genes associated with DNA damage are mutations (mt) and overexpression (↑) of p53 and hypermethylation (me) of MLH1 and MGMT. Lesions associated with proliferation as overexpression of EGFR, CyclinD1, STAT3, NF-κB, downregulation (↓) of p27, and mutations in EGFR, HRAS, and PI3KCA, and hypermethylation of p16 and DAPK. Mutations and consequent overexpression of p53 and upregulation of Bcl-2 lead to loss of apoptotic regulation. Mutations in NOTCH1 and overexpression of VEGF and COX-2 lead to angiogenesis. Invasion is governed by overexpression of COX-2 and MMP and hypermethylation and consequent downregulation of E-cadherin. DAPK, death-associated protein kinase; EGFR, epidermal growth factor receptor; VEGF, vascular endothelial growth factor;↑, upregulation;↓, down regulation.

Molecular landscape of oral cancer development. The primary sequential histopathology of normal squamous mucosa through severe epithelial dysplasia, invasive squamous cell carcinoma, and metastatic tumor deposit in the lymph node and molecular lesions are outlined emphasizing the critical genes and aberrations as overexpression (↑), downregulation (↓), mutations (mt), and hypermethylation (me). The major attributable risk factors of tobacco, areca nut, alcohol, and HPV16/18 are indicated. [Modified from Fletcher (2013), Lippman and Hong (2002)]. HPV, human papillomavirus.
Califano et al. (1996) highlights molecular pathogenesis at the chromosomal level in the regions encompassing 9p loss (p16) toward initiation of precursor lesions; 3p (FHIT) and 17p (p53) in dysplasia's; 11q (Cyclin D1), 13q (Rb), and 14q (microsatellite markers—D14S81, D14S510) with progression to carcinoma in situ; and the invasive oral cancer profile with aberrations of chromosomes 6p (microsatellite marker and TCTE gene), 8p (microsatellite marker), and 4q (microsatellite marker and FABP2). Furthermore, the theme of simultaneous or sequential involvement of molecular aberrations at various stages toward progression of oral cancers has been elaborated in several subsequent reports (Gollin, 2014; Pérez-Sayáns et al., 2009; Pickering et al., 2013).
In addition, literature on the molecular pathogenesis of oral cancers has identified a multitude of potential biomarkers, including Cyclin D1, MKI67, CDKN2A, CCND1, and RB1 associated with cell cycle; EGFR, PI3KCA, Ki-67, MYC, and SERPINB3 associated with cell proliferation; E-cadherin, MMP, VCAN, and MUC4 associated with cell adhesion; VEGFR, COX-2, HMOX1, and CXCL8 associated with angiogenesis; and TP53, p21, Bcl-2, Survivin, FAS, and FASLG associated with apoptosis (Pérez-Sayáns et al., 2009; Ram et al., 2011; Rivera and Venegas, 2014; Rivera et al., 2017; Sinevici and O'sullivan, 2016; Stransky et al., 2011).
The identification of known and novel tumor markers, including somatic mutations, deregulated expression, epigenetic regulation, and single nucleotide polymorphisms (SNPs) in oncogenes and tumor suppressor genes may have far-reaching implications to accurately predict the biologic behavior of the tumor at any given stage and its response to therapy. Hence, the focus of our review is to understand oral biology and indicate an overall integrative concept of the applications of molecular biomarkers as predictive, prognostic, and drug response targets. The molecular aberrations identified in the current review were observed in at least one-third (30%) of the oral cancer patients, thus providing higher potential of translation to the clinic. The imminent translation of the cumulative data has implications in better oral cancer patient management with significant improvement in prognosis and overall survival rates of oral cancer patients.
Gazing Back, Looking Forward on Molecular Landscapes
Molecular lesions in specific pathways are responsible for reprogramming the cell from a normal phenotype to oral cancer. Genomic data available in oral cancers have led to novel insights into genotype/phenotype correlations through linkage of specific molecular lesions to clinicopathology as synopsized in the subsequent sections. A majority of the literature data examined from 1991 to 2017 on clinically relevant biomarkers in oral cancers have been cited. The molecular pathogenesis reported in at least 30% cancer samples and reported in multiple studies was included. The keywords used to search the MEDLINE/PubMed databases included alterations such as somatic mutation, gene expression, DNA methylation, histone modifications, microRNA, and SNPs associated with the clinicopathology.
Somatic mutations in oral cancers
Cancer is a progressive accumulation of somatic mutations providing selective growth advantage to cancer cells. Our emphasis is on candidate genes mutated in greater than 30% of oral cancer patients globally and hence of translation value in oral cancer patients (Table 1).
EGFR, epidermal growth factor receptor.
The p53 tumor suppressor gene is a commonly mutated gene in 41–70% oral cancer patients (Table 1) (Kandoth et al., 2013; Song et al., 2014; Tabatabaeifar et al., 2014; van Houten et al., 2002). The mutations often result in increased expression of the gene in tumor tissues, although the expressed TP53 does not function as a tumor suppressor protein. Besides, the mutated TP53 protein forms a tetramer with normal TP53, neutralizing its function (Chan et al., 2004). Alternately, the mutations result in absence of TP53 expression due to the termination of TP53 peptide as the mutation results in a stop codon (Chan et al., 2004). Molecular assessment of surgical margins using TP53 antibodies indicated patients with high risk for tumor recurrence postsurgery (van Houten et al., 2002). p53 mutations were detected in 11.8% of betel-related and 22.4% in smoking/drinking-related oral cancers (Thongsuksai et al., 2003).
The signal transducing HRAS gene mutations at codons 12, 13, and 61 have been reported in 35–55% oral cancer patients primarily associated with tobacco in Asian and Western countries (Table 1) (Murugan et al., 2012; Saranath et al., 1991). The HRAS mutations resulted in constitutive activation of the gene and consequent downstream signaling. Interestingly in high incidence areas, HRAS mutations were reported in higher proportion (35–55%) of oral cancer cases, whereas in regions with low incidence of oral cancer, HRAS mutations were observed in 0–9% of oral cancer cases (Murugan et al., 2012). A majority of the HRAS mutations were reported from Asian populations associated with tobacco, areca nut, and smoking habits (Murugan et al., 2012). HRAS is critical in initiating and maintaining the tumor, and thus, disruption of the constitutive activation of HRAS is a novel therapeutic approach in oral cancers.
NOTCH1, EGFR, and PI3KCA are signal transducing transmembrane proteins with a critical role in intercellular communication and have been associated with oral cancers. NOTCH1 signaling regulates VEGF receptors and Ephrin B2 genes involved in angiogenesis (Kofler et al., 2011) and is associated with regulation and development of stem cells and progenitor cells in hematopoiesis (Wilson and Radtke, 2006). However, next generation sequencing has demonstrated NOTCH1 mutations in 54% oral cancers and 60% preneoplastic lesions in Chinese patients indicating a role in oral cancer progression (Izumchenko et al., 2015). Song et al. (2014) demonstrated 43% mutation in NOTCH1 gene, associated with overall survival of the patients. A majority of the NOTCH1 mutations are clustered around “ligand-binding” domain leading to increased activity associated with cancer (Arnett et al., 2010).
Epidermal growth factor receptor (EGFR) is a critical transmembrane receptor protein deregulated in several carcinomas. Several studies in Spanish and Korean oral cancer patients demonstrated about 30% mutations in EGFR (Table 1) (Hsieh et al., 2011; Lemos-González et al., 2007). Overexpression of EGFR with/without identified mutations was observed in >40% oral cancer patients (Table 2) (Gröbe et al., 2014; Gupta et al., 2015; Mehta et al., 2015; Rautava et al., 2008; Saranath et al., 1993; Shiraki et al., 2005; Solomon et al., 2016). Genes for downstream effectors of EGFR, including KRAS, MAPK1, and Cyclin D1, were also amplified or mutated in oral cancers. Differential EGFR expression in oral cancer patients was observed with differential sensitivity to antitumor effects of EGFR inhibitors. Thus, functional genomic analysis identified EGFR activation as a common genetic event with clinical implications as theranostic biomarker in oral cancers (Sheu et al., 2009).
+, upregulation; −, downregulation; VEGF, vascular endothelial growth factor.
Furthermore, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit (PIK3CA) mutations E542K, E545K, and H1047R with high oncogenic potential were observed in 30.5% patients with advanced stages of cancer and were associated with metastasis (Kozaki et al., 2006; Lui et al., 2013). Besides, the prognostic implication of PIK3CA mutant tumors demonstrated increased sensitivity to treatment with mTOR/PI3K inhibitor (BEZ-235) as demonstrated in in vivo mouse model systems (Lui et al., 2013).
In general, the somatic mutations in oral cancers result in overexpression of oncogenes and/or downregulation of tumor suppressor genes culminating in uncontrolled cell proliferation and reduced apoptosis. The specific mutations leading to consequent deregulated protein may be useful as novel theranostic biomarkers in association with personalized targeted therapy using small inhibitory molecules identified from chemical database libraries using structure-based and pharmacophore-based in silico approaches, which utilize the knowledge about the receptor and ligand, and are an established computational tool in in silico drug design. Computational approaches in target identification and drug discovery are a relatively novel domain in disease therapeutics with availability of state-of-the-art databases. Thus, genomic alterations can be targeted through structure based and pharmacophore based modeling in several diseases, including cancer (Katsila et al., 2016; Maurer et al., 2012).
Discovery of a binding pocket on Ras genes with functional significance provided novel approaches to defining therapeutic inhibitors. The small-drug like molecules need to be validated on in vitro oral cancer cell lines and in vivo animal model systems (Maurer et al., 2012). Besides being inhibitors to downstream targets of Ras pathway, MEK and mTOR may prove effective in oral cancer patients with HRAS mutations (Kiessling et al., 2015).
The strategies used in Ras-associated cancers may be used for targeting several other driver genes of oral carcinogenesis. Monoclonal antibodies and inhibitory molecules to EGFR gene are in use in oral cancer patients, elaborated in subsequent section. The targeted small molecule inhibitors of additional genes, including p53, PI3KCA, and ATR-CHEK1, may also prove therapeutically effective with these genes upregulated in oral cancers (Aung and Siu, 2016; Boeckler et al., 2008; Gkeka et al., 2012, 2013; Sankunny et al., 2014). The high specificity of the small molecule inhibitors will reduce adverse effects seen in conventional cytotoxic drugs. The advent of in silico drug discovery has established a promising technique for personalized drug design and development.
Deregulated transcriptional profiles in oral cancers
Somatic mutations and epigenetic modifications in oral cancer cells result in either upregulation or downregulation of the gene transcripts affecting protein expression, thus identifying specific biomarkers of clinical significance in oral cancers. The genes deregulated in more than 30% oral cancer patients with potential use as clinical biomarkers are indicated in Table 2 and highlighted below.
Cyclin D1, a key regulatory protein at G1/S checkpoint of the cell cycle, is overexpressed in 39–71% of oral cancer patients (Table 2) (Angadi and Krishnapillai, 2007; Das et al., 2011; Shiraki et al., 2005; Zhao et al., 2014). Furthermore, a meta-analysis indicated association of cyclin D1 overexpression with tumor size, nodal metastasis, advanced clinical stages, and poor survival in Asian oral cancer patients from India, China, and Japan (Zhao et al., 2014).
Ki-67 protein associated with initiation of DNA replication reflects proliferative potential of oral cancers. Ki-67 expression quantitated immunohistochemically showed overexpression in 45–100% oral cancer cases and correlated with high tumor grade and overall survival (Freudlsperger et al., 2012; Kurokawa et al., 2005; Watanabe et al., 2010). The increased expression of Ki-67 associated with increased cell proliferation, demonstrated better response to chemotherapy and radiotherapy, and paradoxically with poor prognosis and poor overall survival in oral cancers (Couture et al., 2002; Freudlsperger et al., 2012; Myoung et al., 2006; Watanabe et al., 2010; Xie et al., 2016).
EGFR, a tyrosine kinase receptor, expressed in basal keratinocytes of the oral epithelium, is upregulated in 39–95% oral cancer patients and associated with poor prognosis (Table 2) (Gröbe et al., 2014; Gupta et al., 2015; Mehta et al., 2015; Rautava et al., 2008; Saranath et al., 1993; Shiraki et al., 2005; Solomon et al., 2016). An increase in EGFR expression was observed in 50% dysplasias, indicating EGFR deregulation as an early event in oral cancers, and EGFR as a predictive biomarker identifying dysplasias with higher risk to progress to a malignant phenotype (Rautava et al., 2008). The increased expression and activity of EGFR are associated with tumor proliferation, invasion, and metastasis (Doumiati et al., 2012; Lin et al., 2014). Positive EGFR tumors were associated with poor prognosis and poor response to chemotherapy (Aquino et al., 2012).
Several theranostic strategies are developed to inactivate the EGFR pathway, including monoclonal antibodies against the extracellular domain of EGFR (Hiraishi et al., 2008). Currently, the only targeted therapy in head and neck cancers, including oral cancers, is Cetuximab (Erbitux™), a monoclonal antibody against EGFR, approved by US-FDA in 2006, and useful in patients with persistent oral lesions with high risk dysplasia and oral cancers with deregulated EGFR (Cassell and Grandis, 2010). The promising preclinical and early clinical results in advanced head and neck cancer patients treated with radiation were compared with radiation plus Cetuximab (54 months vs. 28 months) in which EGFR expression emerges an independent prognostic determinant for locoregional disease control and overall disease-free survival (Harari and Huang, 2006).
However, this treatment is potentially toxic, and established criteria are unavailable to distinguish responsive patients from nonresponders (Lord et al., 2008). Several additional targeted molecules against EGFR include tyrosine kinase inhibitors—Erlotinib, Gefitinib, and Lapatinib, and monoclonal antibodies—Panitumumab, Zalutumumab, and Nimotuzumab, which are potential drugs for treatment of oral cancers (Fung and Grandis, 2010).
Vascular endothelial growth factor (VEGF) induces neovascularization and is critical in cancer tissues particularly as the tumor increases in size, and consequently facilitates metastasis (Hanahan and Weinberg, 2011). Deregulation of VEGF was demonstrated in 41–87% oral cancers with increase in VEGF associated with the depth of tumor invasion and poor prognosis (Table 2) (Arora et al., 2005; Johnstone and Logan, 2007; Kim et al., 2015; Smith et al., 2000).
NF-κB (nuclear factor kappa B) is involved in several cellular processes, including inflammation, transformation, proliferation, angiogenesis, invasion, metastasis, chemoresistance, and radioresistance (Chaturvedi et al., 2011). NF-κB was found to be upregulated in 50–63% oral cancer patients (Table 2) (Bindhu et al., 2006; Piva et al., 2013). Activation of NF-κB was observed during oral cancer progression and having an impact on its metastatic potential and poor survival (Tang et al., 2017). NF-κB has been found to stimulate the expression of a number of pro-inflammatory cytokines such as TNF-α and interleukin–1 and −6 (IL-1 and IL-6), as well as the degradative enzymes such as MMPs (Bindhu et al., 2006).
Bindhu et al. (2006) provided evidence that NF-κB progresses through premalignant phase to invasive phase of oral cancer, indicating an early event in malignant transformation of oral cancers. Activated NF-κB binds to specific target genes, including COX-2, Cyclin D1, Bcl-2, Bcl-xL, Survivin, and XIAP, which mediate the process of chemoresistance and radioresistance (Li and Sethi, 2010). Given the tumor-promoting role of NF-κB, targeting NF-κB for tumor prevention and therapy might be beneficial.
COX-2 (cyclooxygenase-2) has been implicated in angiogenesis and cancer invasion. COX-2 upregulation was observed in 68.4–80% oral cancer patients (Table 2) (Aruldoss et al., 2016; Li and Cui, 2013). Increased expression of COX-2 results in angiogenesis through elevated levels of prostaglandins, E2 and VEGF, leading to tumor promotion and progression (Polanska et al., 2014). COX-2 has been implicated to mediate radioresistance in various tumors, including oral cancers (Terakado et al., 2004).
Constitutive activation of STAT3 (signal transducer and activator of transcription 3) in head and neck cancers is accompanied by increase in STAT3 phosphorylation and linked to cell proliferation, differentiation, and apoptosis (Leeman et al., 2006). Aberrant activation of TGF/EGFR has been associated with constitutive activation of STAT3 in head and neck cancers (Bromberg, 2002). Upregulation of STAT3 was found in 42–62% oral cancer patients (Table 2) (Grandis et al., 2000; Shah et al., 2005).
The p53 gene primarily associated with apoptosis, cell cycle arrest, and DNA repair is mutated in several cancers with consequent overexpression of the gene. Immunohistochemical analysis of TP53 indicated overexpression in 37–75.8% oral cancers (Table 2) (Gupta et al., 2015; Kurokawa et al., 2005; Saranath et al., 1999; Shiraki et al., 2005; Siegelmann-Danieli et al., 2005; Stoll et al., 2000). TP53 deregulation is associated with resistance to cisplatin and 5-fluorouracil (5-FU) neoadjuvant chemotherapeutics in oral cancers and hence an effective biomarker predicting therapeutic response (Adelstein et al., 2003; Kanayasu et al., 2004; Perrone et al., 2010).
Bcl-2 is an antiapoptotic gene with overexpression in 56–87% oral cancers and overexpression associated with nodal status and survival (Table 2) (Camisasca et al., 2009; Teni et al., 2002). Besides, 16% oral lesions show Bcl-2 overexpression implying potential as a predictive biomarker (Teni et al. 2002). In vitro studies indicate association of Bcl-2 overexpression with resistance to cisplatin based drugs (Xiong et al., 2016).
Tumor invasion and metastasis involve increased expressions of MMPs, especially MMP2 and MMP9 because they degrade type IV collagen, which is an important component of extracellular matrix (Egeblad and Werb, 2002). Both MMP2 and MMP9 have been demonstrated to play a critical role in the process of invasion and metastasis of oral cancers. Upregulation of MMP2 was observed in 71–86.7% oral cancer patients, while MMP9 was upregulated in 58.5–90% oral cancer patients and associated with lymph node metastasis (Table 2) (de Vicente et al., 2005; He et al., 2016; Katayama and Bandoh, 2004; Monteiro et al., 2016; Zhou et al., 2010).
Downregulated genes in more than 30% oral cancer patients include E-cadherin and p27. E-cadherin, a cell adhesion molecule, plays a key role in development of invasive and recurrent carcinoma. The balance between the proportion of E-Cadherin and N-Cadherin is a hallmark of epithelial–mesenchymal transition with decreased E-cadherin and increased N-cadherin favoring the transition to mesenchymal characteristics and invasiveness of the tumor leading to metastasis (Maeda et al., 2005).
Downregulation of E-cadherin expression was observed in 33–62.5% oral cancer patients (Table 2), associated with tumor site and mortality (Fan et al., 2013; von Zeidler et al., 2014). Loss of E-cadherin was observed in high risk leukoplakia indicating progression to oral cancers (von Zeidler et al., 2014). Reduced expression of p27 was reported in 29–84% oral cancers (Table 2), 82% invasive oral cancers, and 39–70% severe dysplasias (Fillies et al., 2007; Jordan et al., 1998; Kudo et al., 2001; Schoelch et al., 1999) indicating deregulation of p27 as an early event and in progression of oral carcinogenesis.
Several classes of small molecule inhibitors targeted toward deregulated proteins in conjunction with chemotherapy and radiotherapy are under investigation. These include kinase inhibitors, inhibitory peptides, antisense RNA, and proteasome inhibitors that can block activation of oncogenic proteins. 3-{[1-(3-Chloro-4-fluorophenyl)-3,5-dioxo-4-pyrazolidinylidene]methyl}-phenyl-2-thiophene carboxylate was identified as a potent inhibitor of EGFR kinase using in silico technology and in vitro experimentation (Li et al., 2012).
Several chemotherapeutic agents, including Paclitaxel, Vinblastine, Vincristine, Doxorubicin, Daunomycin, 5-Fluorouracil, Cisplatin, Tamoxifen, and Bortezomib, have been reported to induce NF-κB activation (Li and Sethi, 2010). Paclitaxel was found to transiently induce NF-κB activity through PI3K cascade and in combination with NF-κB inhibitor (BAY 11-7085) could increase the therapeutic efficacy (Mabuchi et al., 2004). BH3-mimetic compounds offer a novel approach for treating chemoresistant cancers by blocking select pro-survival Bcl-2 family members. The potential of BH3 mimetics in cancer therapy was demonstrated by ABT-737 (Oltersdorf et al., 2005) and its orally available clinical derivative ABT-263 (Navitoclax), currently in phase 2 clinical trial (Delbridge and Strasser, 2015; Tse et al., 2008).
Preclinical data suggest that combination of Navitoclax with conventional chemotherapeutics may increase efficacy in solid tumors (Tan et al., 2011). A STAT3 selective decoy oligonucleotide inhibitor has demonstrated biologic activity in head and neck patients in phase 0 clinical trial (Sen et al., 2012). WP1066 demonstrated antitumor effects by suppression of JAK2-STAT3 signaling in head and neck cancer models (Kupferman et al., 2009). FLLL32, a novel small molecule STAT3 inhibitor derived from curcumin, sensitized head and neck cancer cells to cisplatin treatment in vitro (Abuzeid et al., 2011).
Epigenetic regulation in oral cancers
Epigenetic regulation, including DNA methylation, histone modification, and chromosomal remodeling, and deregulation of microRNAs play an important role in oral cancer. Transcriptional silencing of genes in oral cancer through hypermethylation of CpG islands in the promoter is often an early event in oral cancer and thus indicate preventive biomarkers and result in downgrading oral cancer in regions with high incidence of oral cancer (D'Souza and Saranath, 2015; Hashimoto et al., 2012; Huang et al., 2009; Taioli et al., 2009; Xing et al., 2013). Besides, the ease of analysis of methylated CpGs using methylation specific polymerase chain reaction (MS-PCR) and nucleotide sequencing with high specificity, sensitivity, and quicker turnaround time of the assay will facilitate clinical applications. The density of methylated CpGs in specific genes, including death-associated protein kinase (DAPK), O-6-methylguanine DNA methyltransferase (MGMT) and p16 in long-term tobacco users, premalignant lesions, tumor adjacent mucosa, and oral cancer, corroborates the predictive value of these biomarkers (Kulkarni and Saranath, 2004).
A meta-analysis identified 7 genes hypermethylated in 34–79% oral cancers (Table 3) (D'Souza and Saranath, 2015). Silencing of specific genes by hypermethylation has been associated with clinipathological features, for example, E-cadherin association with increased metastasis, p16 with prognosis, DAPK, MGMT, MLH1, and FHIT as early event biomarkers (Hashimoto et al., 2012; Huang et al., 2009; Taioli et al., 2009; Xing et al., 2013). Thus, hypermethylation of specific genes is a valuable biomarker in oral cancers and may identify effective therapeutic targets.
Ac, acetylation; Me, hypermethylation; +, upregulation; −, downregulation; DAPK, death-associated protein kinase.
Histone modifications resulting in chromatin remodeling alter gene function, and the deregulation has been associated with cancer prognosis and patient survival (Chen et al., 2013). Cumulative studies on histone modifications in oral cancers demonstrated histone H3 methylation at specific lysine molecules K4 in 39% (n = 145), K9 in 47% (n = 199), and K27 in 45% (n = 160) oral cancers, and acetylation at K4 in 37% (n = 171), K9 in 80% (n = 10), and K18 in 39% (n = 152) oral cancers (Table 3) (D'Souza and Saranath, 2015). Besides histone tail modifications, histone deacetylase 6 (HDAC6) enzyme was overexpressed in 74% oral cancers in correlation with tumor aggressiveness (Sakuma et al., 2006).
Several microRNAs (miR) are deregulated in oral cancers, with either tumor suppression or oncogenic role regulating cell proliferation, differentiation, migration, apoptosis, and signal transduction in oral carcinogenesis. miR-31, miR-21, miR-146a, miR-211, miR-204, miR-24, and miR-155 are upregulated in 70% oral cancers; and miR-125b, miR-145, miR-126, miR-203, miR-218, miR-585, miR-99a, and miR-137 were downregulated in 69% oral cancers, in association with metastasis, poor prognosis, and disease-free survival (Table 3) (D'Souza and Saranath, 2015).
In contrast, miR-200 was epigenetically activated in oral cancers by hypomethylation (Wiklund et al., 2011). The differential expression of microRNAs in cancer and normal buccal mucosa provides novel biomarkers in oral cancer prognosis and targets for potential therapy. The identification of metastamir, a family of microRNAs with pro-metastasis (miR-10b, miR-373, miR-520c, miR-21, miR-143, and miR-182) and anti-metastasis (miR-335, miR-206, miR-146a/b, and miR-31) activities, indicates new therapeutic strategies (Hurst et al., 2009).
The precise epigenetic modifications, including DNA hypermethylation, histone modification, and microRNA profile, will facilitate informed decision on use of DNA methyltransferase inhibitors (DNMTi)—Azacytidine and Decitabine, and histone deacetylase inhibitor (HDACi)—Trichostatin A, Valproic acid, and Suberoylanilide hydroxamic acid (SAHA). In addition to epidrugs, miRNA antisense oligonucleotides may be used in an attempt to restore normal functions disrupted by deregulated miRNAs. The novel therapeutic agents may be used as stand-alone drugs or as combination therapy in oral cancers.
Genomic variants in oral cancers
A small proportion of high risk individuals with tobacco/areca nut/alcohol develops oral cancer and is distinguished by the differential inherent genomic constitution. Thus, the genomic constitution of an individual plays a critical role in oral cancer susceptibility. The genomic variants as SNPs are located in exonic or intronic regions of the genes, although a majority of the SNPs are observed in the noncoding introns (Zhao et al., 2003). SNPs result due to genetic drift, gene flow, or migration and natural stabilizing/directional/disruptive selection (Li and Leal, 2009). SNPs have been associated with several cancers, including prostate, breast, ovarian, and colorectal cancers in association with increased/decreased risk, prognosis, and response to treatment (Glinskii et al., 2009).
Several studies on SNP association with oral cancers report a wide range (9–90%) of oral cancer patients containing the specific SNP in association with increased or decreased susceptibility to the cancer. In the current review, we have included SNP associations indicating ≥50% increased or decreased risk, to identify predictive and prognostic biomarkers. The SNPs in DNA repair gene MLH1 (rs1800734) indicated highest risk to oral cancer (OR: 6.73; 95% CI: 3.9–11.5) for the homozygous SNP genotype (Jha et al., 2013).
Besides, SNPs in genes associated with apoptosis, including Survivin (rs2071214) and AKT1 (rs189037), cell cycle- p27 (rs34329) and ATM (rs189037), immune function- IL-1β (rs16944) and IL-18 (rs187238), cell communication- CD44 (rs187115), cell proliferation- TGF-β (rs1982073), PREX2 (rs4512367), and GRIK2 (rs1335022), and xenobiotic metabolism- ADH1B (rs1229984) and CYP1A1 (rs4646903), demonstrated increased risk in oral cancer (Table 4). High throughput microarray and next generation sequencing studies in oral cancer patients identified 26–93 SNPs in genes associated with signal transduction, transcription factors, cell migration, and cell metabolism, and association with carcinogenic agents in tobacco and areca nut (Bhatnagar et al., 2012; Yadav et al., 2014).
SNP, single nucleotide polymorphism; +, increased risk; −, decreased risk.
Several studies emphasize the importance of SNP-oral cancer risk association, and a recent meta-analysis has highlighted 34 SNPs in association with increased risk to oral cancers (Multani and Saranath, 2016). The SNPs associated with increased risk of oral cancer in multiple studies and varied populations were rs1800471 (TGF-β), rs1048943 (CYP1A1), rs1800870 (IL-10), rs11549467 (HIF), and rs861539 (XRCC3), whereas association of decreased risk was observed with homozygous WT genotype of the SNPs in TGF-β, CYP1A1, IL-10, HIF, and XRCC3 (Multani and Saranath, 2016). In contrast, the homozygous WT genotypes in rs1801133 (MTHFR) and rs20417 (COX-2) demonstrated an increased risk, and the corresponding homozygous SNP genotypes were associated with decreased risk to oral cancer (Multani and Saranath, 2016). The meta-analysis provided cumulative data on a number of SNPs associated with oral cancer risk in various ethnic populations (Multani and Saranath, 2016).
A meta-analysis of 2186 cases and 4488 controls revealed SNP in ADH1B (rs1229984, Arg47His) with homozygous WT genotype Arg/Arg and increased risk of oral cancer in Asian cohorts in alcohol drinkers versus nondrinkers and correlated with increased recurrence and poor survival (Peters et al., 2004). Furthermore, a meta-analysis of 13 studies, including 1468 cases and 2183 controls, identified a significant increase in oral cancer risk for the homozygous SNP in CYP1A1 (rs1048943, Ile462Val) (Yang et al., 2015). SNPs may thus provide useful adjuncts in risk predisposition to oral cancer and may aid in implementation of aggressive tobacco cessation programs.
Genetic testing must be implemented as a part of the clinical management of families harboring the high risk SNPs. As observed in oral cancers, a large number of high risk SNPs exhibit modest disease association when analyzed individually, whereas as a gene panel, a polygenic disease model will explain a significantly higher risk of the SNP to the disease. A recent study evaluated the risk stratification ability of 77 SNP polygenic model for breast cancer and identified that women in the top 10% of genetic risk would reach this risk threshold in their early 30s, aiding in better patient management and effective screening (Mavaddat et al., 2015).
Similarly, a colorectal cancer study investigated 27 SNPs in association with clinicopathology and family history (Hsu et al., 2015). The study identified that the risk determined by the 27 SNP polygenic model in combination with family history can have a substantial impact on age at which individuals will be at high risk of colorectal cancer. Undergoing screening using the polygenic model will have far reaching benefits of early cancer detection.
Besides identifying individuals with high risk to oral cancers, SNPs also influence DNA methylation pattern. The presence of SNPs in promoter or enhancer regions also regulates methylation pattern across the genomic region (Smith et al., 2014) as reported in breast cancer, SNP rs2380205 influencing DNA methylation of F-box protein, helicase, 18 (FBXO18); rs2736100 and telomerase reverse transcriptase (TERT) in lung adenocarcinoma; and rs16906252 and MGMT in glioblastoma (Heyn et al. 2014; McDonald et al., 2013). However, this has not been reported in oral cancer.
Theranostic Biomarkers in Oral Cancers
The current conventional modalities with technological advances and combinatorial approaches in surgery, radiation, and chemotherapy are associated with systemic toxicities and adverse side effects and reduce patient compliance (Patel et al., 2008). A significant improvement with increased duration of overall survival in oral cancer patients has not been observed. The strategy of using theranostic biomarkers with biological agents, including small molecular inhibitors and monoclonal antibodies after stratification of patients based on specific molecular biomarkers, may show better results. The oral carcinomas with EGFR positivity are treated with Cetuximab, a monoclonal antibody against EGFR approved by US-FDA in 2006 (Fung and Grandis, 2010).
In addition, Nivolumab, a monoclonal antibody, inhibits programmed cell death-1 protein (PD-1) and blocks PD-1 from interacting with the ligands. Nivolumab has been approved by US-FDA in 2016 for recurrent head and neck cancers, and anti PD-1 antibody Brolizumab (MK-3475) is currently in phase IB clinical trials (Seiwert et al., 2014). Targeting VEGFR a potent angiogenic molecule reduces endothelial migration. Bevacizumab, a humanized monoclonal antibody that inhibits VEGF, is in phase II trial in combination with Cetuximab in head and neck cancers (ClinicalTrials.gov; Karamouzis et al. 2007). Adenovirus ONYX-015 restores TP53 function eventually causing apoptosis. ONYX-015, an adenovirus modified selectively to replicate in and kill cells that harbour p53 mutation while normal cells are unaffected, is a distinct treatment possibility as p53 mutation is observed in 41–70% oral cancers.
Currently, Ad5CMV-p53 (virally mediated transduction of p53) has completed phase I/II clinical trial (ClinicalTrials.gov Identifier: NCT00064103) in patients with premalignant carcinoma of the oral cavity. Flavopiridol (cyclin-dependant kinase inhibitor) has completed phase II clinical trial (ClinicalTrials.gov Identifier: NCT00020189) in metastatic head and neck cancers, and PS-341 (Bortezomib) a proteasome inhibitor has completed phase II clinical trial (ClinicalTrials.gov Identifier: NCT00103259) in treating patients with locally recurrent or metastatic head and neck cancers (ClinicalTrials.gov).
The molecular landscape in oral cancers indicating somatic mutations in p53, HRAS, NOTCH1, EGFR, and PI3KCA; consequent deregulated expression of Cyclin D1, Ki-67, EGFR, VEGF, p53, Bcl-2, E-cadherin, and p27; and DNA methylation in p16, p15, DAPK, E-cadherin, MGMT, MLH1, and FHIT resulting in deregulation of the genes can be used as potential targets for effective targeted therapy with anti-sense oligonucleotide and monoclonal antibodies, and small molecule inhibitors are an approach for better patient management in oral cancer. Integration of genomics and functional approaches in a clinical setting holds promise.
Conclusions and Outlook
Molecular profiling has led to the understanding of cancer biology with consequent identification of biomarkers relevant to development and progression of normal cells to a malignant phenotype. The biomarkers may be used in clinical practice as early event, as well as risk biomarkers, biomarkers in response to treatment, and prognostic biomarkers in cancer patients identifying disease-free survival and risk of metastasis.
Furthermore, the biomarkers may identify therapeutic targets with stratification of patients based on the specific target to positively impact morbidity and mortality. Although there are a number of reviews regarding oral cancer biomarkers, none of them currently provides a general overview of the topic. In the current review, literature cited from 1991 to 2017 in MEDLINE/PubMed databases was used to search for biomarkers for early event, risk, prognosis, and theranostics using the following keywords: somatic mutations, gene expression, DNA methylation, histone modifications, microRNA, and SNPs associated with clinicopathology. The pathogenesis using the markers was reported minimally in 30% cancer samples and identified in multiple studies.
The molecular aberrations identified were somatic mutations in p53, HRAS, NOTCH1, EGFR, and PI3KCA; upregulation of Cyclin D1, Ki-67, EGFR, VEGF, NF-κB, COX-2, STAT3, p53, MMP-2, MMP-9, Bcl-2; downregulation of E-cadherin and p27; epigenetic modifications with DNA hypermethylation in p16, p15, DAPK, E-cadherin, MGMT, MLH1, and FHIT; and histone modifications and deregulation of specific microRNAs in critical cellular pathways leading to progression of oral cancers (Fig. 1). Molecular biomarkers indicative of prognosis were mutations in p53, NOTCH1, and EGFR; deregulated expression of Cyclin D1, Ki-67, EGFR, VEGF, NF-κB, and Bcl-2; and hypermethylation of p16, histone modifications, and deregulated microRNAs.
With respect to conversion of normal cells in the oral cavity to malignant phenotype occurring at the transformation rate of 3–8.1%, two lines of thought are indicated. First, the markers of early events such as deregulated expression of EGFR, NF-κB, E-cadherin, and p27 and DNA hypermethylation of DAPK, RUNX3, MGMT, MLH1, and FHIT may be translated to identify individuals at high risk of developing oral cancer (Fig. 2). Second, genomic variants indicating increased risk to oral cancers as represented by the SNPs in MLH1, p27, IL-1β, Survivin, AKT1, CD44, TGF-β, PREX2, ADH1B, IL-18, ATM, CYP1A1, and GRIK2 (Table 4).
The heterogeneity of the identified molecular biomarkers may be due to the differences in ethnicity of the oral cancer patients highlighting the role of population-tailored biomarkers. Oral cancer specific protein biomarkers, S100A9 and IL-6, were identified in the Hungarian population and fibrinogen (alpha/beta/gamma) chain, haptoglobin, leucine-rich alpha-2-glycoprotein, and ribosomal protein S6 kinase alpha-3 (RSK2) in the Taiwanese population (Csősz et al., 2017; Yu et al., 2016), while SNPs rs11130760 (FHIT), rs1957358 (SAMD4A), rs4512367 (PREX2), rs2124437 (RASGRP3), and rs1335022 (GRIK2) indicating high risk to oral cancer were identified in the Indian population (D'Souza et al., 2017; Multani et al., 2016). Different ethnic groups highlight the significance of population-tailored biomarkers to identify oral cancer specific molecular alterations for cost-effective screening purposes.
Aggressive follow-up facilitating tobacco cessation and diagnosis at early stages of the disease will decrease oral cancer incidence and result in downgrading the cancer in the population. Besides, the molecular landscape may result in defining targets for drug development using advanced in silico structure-based and pharmacophore-based approaches. Theranostic biomarkers with/without companion diagnostics are currently cost-ineffective unless covered by the patient's medical insurance. Thus, cost-effective interventions using novel biomarkers as defined by the genomic fingerprint need to be developed for better oral cancer patient management. Oral saliva, blood, urine, and other body fluids represent a significant source of noninvasive biomarkers substituting invasive and costly procedures like biopsies, to evaluate the condition of both patients and healthy individuals (Al-Tarawneh et al., 2011).
Furthermore, the availability of high-throughput DNA sequencing technology together with DNA hybridization, genomic fingerprinting, and 16S rRNA gene cloning has made it feasible to identify molecular lesions in primary tumors and body fluids (Horváth et al., 2017). Molecular biomarkers and next-generation diagnostics provide tremendous opportunity to delivering noninvasive, cost effective, and accessible detection and monitoring of oral cancers. The identified molecular lesions may improve diagnostics, treatment, and prevention strategies. Health technology assessment (HTA) should be commenced in dentistry regarding guidelines for the development and future of oral health services (Steuten, 2016).
Footnotes
Acknowledgment
The authors acknowledge Department of Science and Technology, Government of India, New Delhi, India, for the INSPIRE Senior Research Fellowship to Ms. Wendy D'Souza.
Author Disclosure Statement
The author(s) declare(s) that no conflicting financial interests exist.