
Abstract
Abstract
The cell surface proteome of the foodborne pathogen Listeria monocytogenes, the etiological agent of listeriosis, is critical for understanding the physiological processes associated with stress resistance and persistence in the environment. In this context, the most widespread mode of growth for bacterial cells in natural and industrial environments is in biofilms. Cell surface proteins are, however, challenging to characterize because of their low abundance and poor solubility. Moreover, cell surface protein extracts are usually contaminated with cytoplasmic proteins that constitute the main signal in proteomic analysis. This study aimed to compare the efficiency of three methods to extract and explore surface proteins of L. monocytogenes growing in a biofilm: trypsin shaving, biotinylation, and cell fractionation. Peptide separation and identification were performed by shotgun proteomics using high-performance liquid chromatography combined with tandem mass spectrometry (LC-MS/MS). The biotinylation method was the most effective in extracting surface proteins, with the lowest rate of contamination by cytoplasmic proteins. Although presenting a higher contamination rate in cytoplasmic proteins, the other two techniques allowed the identification of additional surface proteins. Seven proteins were commonly retrieved by the three methods. The extracted proteins belong to several functional classes, involved in virulence, transport, or metabolic pathways. Finally, the three extraction methods seemed complementary and their combined use improved the exploration of the bacterial surface proteome. These new findings collectively inform future discovery and translational proteomics for clinical, environmental health, and industrial applications.
Introduction
The most widespread mode of growth for bacterial cells in natural and industrial environments is in biofilms. Cell surface proteins are difficult to characterize because of low abundance and poor solubility. Moreover, cell surface protein extracts are usually contaminated with cytoplasmic proteins that constitute the main signal in proteomic analysis.
In this context, bacterial cell surface proteome constitutes the so-called proteosurfaceome of a bacterium (Cullen et al., 2005; Desvaux et al., 2006, 2018). Although the totality of the molecules found at the bacterial cell surface corresponds to the surfaceome, the proteosurfaceome specifically refers to the protein subset of the surfaceome, which can be involved in diversified processes, and contributes to the interface between the bacterium and its environment.
Among bacteria involved in biofilm formation, Listeria monocytogenes is an important foodborne pathogen associated with high mortality rates in large outbreaks (Allerberger and Wagner, 2010). Capable of growing at refrigeration temperatures, in a wide range of pH (4.3–9.6), in salt concentrations up to 10% NaCl and low water activity (Aw down to 0.90), it can contaminate and persist in food-processing environment (Renier et al., 2011). In the context of pathogenesis, surface proteins are key factors to understand invasion mechanisms of host cells and thriving capabilities in stress conditions including hostile niche.
Annotation of L. monocytogenes EGD-e (Glaser et al., 2001) genome predicted 133 surface proteins, accurately classified according to their anchoring systems and potential structural domains (Cabanes et al., 2002). Essential roles for surface proteins include bacterial growth, transport, virulence, sensing of and resistance to environmental conditions, signaling and adhesion to substrates, and intercellular interaction for biofilm formation. These surface proteins correspond to nearly 5% of all the predicted proteins from the complete genome, and interestingly, represent the major difference between L. monocytogenes and the nonpathogenic Listeria innocua, highlighting their potential role in pathogenesis (Cabanes et al., 2002).
Although the involvement of surface proteins in virulence is well documented, there are only few investigations concerning the role of cell surface proteome in other mechanisms like adaptation, resistance, or persistence in the environment. Examining the proteosurfaceome of L. monocytogenes in biofilms is important and timely because it is the predominant mode of growth in food workshops (Giaouris et al., 2014, 2015), thereby influencing the physiology and the cell envelope composition of sessile cells (Azeredo et al., 2017).
This study aimed to compare the efficiency of three methods to extract and explore surface proteins of L. monocytogenes growing in a biofilm: trypsin shaving, biotinylation, and cell fractionation.
Materials and Methods
Strain and culture conditions
L. monocytogenes EGD-e strain, serogroup 1/2a, whose genome was sequenced in 2001 (Glaser et al., 2001), was used throughout this study. Routine preculturing and culturing were carried out in tryptic soy broth (TSB; Difco, Fisher Scientific) at 25°C and 150 rpm. Bacterial growth was monitored by measuring the absorbance at 600 nm (OD600). Precultured cells in stationary phase were used to inoculate cultures to obtain a final OD600 of 0.005. After 6 h of growth, cells were harvested by centrifugation (7500g, 15 min) and resuspended in TSB diluted by 1:5 with sterile water in a volume equal to that of the supernatant collected, reaching an OD600 between 0.6 and 0.7. Seven milliliters of the bacterial suspension was poured on each stainless steel (SS) disk (38.5 cm2), corresponding to an inoculation of 108–109 colony-forming units/cm2.
The SS disks were then placed in a sterile petri dish (55 mm diameter) and incubated at 25°C. Bacterial cells were allowed to adhere onto the disk for 3 h in static mode, before removing the medium to eliminate planktonic cells and adding fresh medium. The disks were then incubated for 21 h to reach biofilm formation. To harvest the biofilm, the medium was removed and adherent cells were detached in 10 mL of tryptone salt (tryptone 0.1%, NaCl 0.85%, pH 7.0) by scraping the SS disk with a sterile spoonbill. Cell adhesion and biofilm formation after 24 h incubation at 25°C were evaluated by cell enumeration. Serial dilutions were plated on tryptic soy agar (Difco, Fisher Scientific) and incubated for 24 h at 37°C.
Enzymatic shaving of surface proteins
The shaving method consists of treating intact cells with proteases in an isotonic solution to release exposed peptides. Biofilm cells were harvested by low-speed centrifugation (1000g, 15 min, 4°C) to prevent cell lysis. The bacterial cell pellet was gently washed twice with 1 mL of ice-cold Tris-buffered saline (20 mM Tris–HCl pH 7.4, 150 mM NaCl). Pellet was resuspended in 1 mL of shaving buffer (20 mM Tris–HCl pH 7.4, 150 mM NaCl, 10 mM CaCl2·6H2O, 1 M
Peptides purification and concentration were carried out using Sep-Pak C18 Plus Short cartridges (Waters), pre-equilibrated in two steps with 65% acetonitrile (ACN)/0.1% trifluoroacetic acid (TFA) and 2% ACN/0.1% TFA. Peptides were loaded onto the cartridges, washed with 2% ACN/0.1% TFA, and eluted with 65% ACN/0.1% TFA. Purified peptides were dried with a speed-vacuum and resuspended in 20 μL of 2% ACN/0.1% TFA.
Biotinylation of bacterial cell surface proteins
In the biotinylation method, intact cells are treated with Sulfo-NHS-SS-Biotin, to which the cell membrane is impermeable. This marker molecule reacts specifically with the ɛ-amino group of lysine residues of surface-exposed proteins. Subsequently, labeled proteins can be separated from nonlabeled proteins by affinity chromatography with neutravidin and then analyzed by liquid chromatography combined with tandem mass spectrometry (LC-MS/MS) (Hempel et al., 2010, 2011; Romero-Saavedra et al., 2014; Voss et al., 2014). Biofilm cells were suspended in 10 mL buffer A (phosphate-buffered saline [PBS], 0.01 mM, pH 8 + 1 mM phenylmethylsulfonyl fluoride). The suspension was transferred into weighted tubes and centrifuged at 4000g for 10 min at room temperature (RT).
The bacterial pellet was washed twice, and the weight of wet cells was calculated. Each 100 mg of cells was resuspended in 300 μL buffer A supplemented with 1.5 mM EZ-Link Sulfo-NHS-SS (Thermo Scientific). Biotinylation was performed for 15 min at RT under gentle agitation. Free biotin was removed by centrifugation at 4000g for 5 min at RT and pellet was washed three times with PBS (0.01 M, pH 8 + 500 mM glycine) to block nonreacted biotin. Cells were resuspended in 500 μL buffer A supplemented with 1% (v/v) Triton X-100 and broken at 4°C by vigorous shaking in a Fastprep-24 cell breaker twice for 20 sec. The cell extracts were centrifuged at 20,000g for 30 min at 4°C to pellet the insoluble material.
Labeled proteins were recovered by affinity chromatography in a Monomeric Neutravidin Resin (Thermo Scientific), with gravity flow, using PBS (pH 8) + 1% Triton X-100 as equilibration and wash buffer. Proteins were eluted with an elution buffer (50 mM ditiotreitol [DTT], 2% sodium dodecyl sulfate [SDS] 5% β-mercaptoethanol, 20% glycerol in 62.5 mM Tris–HCl, pH 6.8). A control without biotin labeling was carried out in parallel with the same protocol.
Cell fractionation
The third method tested in this study is the fairly well-established separation of membrane and cell wall components by cell fractionation. However, extractions from cell fractionation are usually heavily contaminated with cytoplasmic proteins and often give insufficient information regarding surface proteins (Rodriguez-Ortega et al., 2006). Biofilm cells were washed twice in Tris–ethylenediamine tetraacetic acid (EDTA) (TE; 20 mM Tris, 5 mM EDTA, pH 7). Pellet was resuspended in 1 mL TE, and bacterial cells were broken using a cell disrupter (One shot cell disrupter, 1–8 mL, 2.7 kbar max, Constant Systems Ltd., Daventry, United Kingdom) by applying 2.5 kbar pressure.
Insoluble materials containing cell walls were removed by centrifugation (13,000g, 15 min, 4°C) and the supernatant was ultracentrifuged (200,000g, 1 h, 4°C). The pellet containing membranes was washed twice in 1 mL Tris 40 mM, pH 8.5. Membranes were suspended in denaturing buffer (1% SDS, 0.1 M DTT, 20 mM Tris–HCl, pH 7.5) before heat treatment (5 min, 95°C). Membrane and cell wall protein extracts were suspended in 100 mM ammonium bicarbonate pH 7.5. Proteins were reduced with 2.5 mM DTT at 56°C for 30 min, alkylated with 25 mM iodoacetamide during 20 min in the dark, and after digested overnight at 37°C with 20 μg of trypsin (Promega) per sample. Extracted peptides were purified and concentrated with Sep-Pak C18 Plus Short cartridges as described previously.
LC-MS/MS and bioinformatic analyses
Each of the three methods described above was applied to three independent biofilm cultures to obtain triplicate protein samples. Samples were loaded onto SDS–polyacrylamide gel electrophoresis gels and subjected to a short electrophoresis to concentrate the soluble proteome in one single band in the first few millimeters of the resolution gel. Excised bands were washed in 25 mM ammonium bicarbonate with 5% ACN for 30 min and twice in 25 mM ammonium bicarbonate with 50% acetonitrile for 30 min. Reduction and alkylation reactions were performed with 10 mM DTT and 55 mM iodoacetamide solutions, respectively, and all bands were finally dehydrated with 100% acetonitrile. The samples were hydrolyzed overnight with 600 ng trypsin and peptides extracted from the gel bands with 100% acetonitrile.
All peptide mixtures were analyzed by nano-LC-MS/MS (Thermo Fisher Scientific) using an Ultimate 3000 system coupled to a Linear Trap Quadropole (LTQ) Velos mass spectrometer (MS) with a nano ion source. For each sample, 8 μL of peptide mixture were first preconcentrated on a C18 precolumn (300 μm inner diameter ×5 cm length) equilibrated with TFA 0.05% in water at 30 μL/min. After 6 min of desalting, the precolumn was switched online with the analytical C18 column (75 μm inner diameter ×25 cm length; 2 μm, Acclaim C18 Pepmap RSLC) equilibrated in 96% solvent A (99.5% H2O, 0.5% formic acid) and 4% solvent B (80%ACN, 19.5% H2O, 0.5% formic acid). Peptides were eluted using 4–55% gradient of solvent B for 50 min at 400 nL/min flow rate.
Eluates were electrosprayed in positive-ion mode at 2.7 kV through a nanoelectrospray ion source heated to 275°C. The LTQ Velos MS was used in collision-induced dissociation top 10 mode (i.e., 1 full scan MS and the 10 major peaks in the full scan were selected for MS/MS). For raw data processing, Thermo Proteome Discoverer v1.2 was used with Mascot v2.3 for database search (www.matrixscience.com). The following parameters were considered for the searches: precursor mass tolerance of 1.5 Da and fragment mass tolerance of 0.5 Da, a maximum of two missed cleavage sites of trypsin, carbamidomethylation, and oxidation set as variable modifications.
Protein identification was validated when at least two peptides originating from one protein showed statistically significant identification Mascot scores >13 (p < 0.05). Ions score is −10 log(P), where P is the probability that the observed match is a random event. Individual ions scores >13 indicate identity or extensive homology. Interrogations were performed against a custom database containing distinct entries corresponding to raw protein sequences and the different predicted mature proteins of L. monocytogenes EGD-e (i.e., DB-Mature-LmoEGDe v2.0, 5838 sequences) based on putative cleavage sites of the signal peptide (SP) (Renier et al., 2015).
The subcellular location of identified proteins was determined with the rational secretomics-based strategy for genomic and proteomic analyses developed by Renier et al. (2012). In brief, the flowchart and decision trees designed to predict the subcellular localization use available bioinformatic tools and mimic the biology of protein secretion taking into consideration successively the presence or not of an SP, the type of SP, the secretion system pathway, the export of proteins lacking an SP, the transmembrane domains (TMD), and conserved cell wall domains.
Results
Global results on proteins identified by the three methods
The data included in our results concerned protein identified with at least two unique peptides to ensure MS/MS results and in at least two biological replicates to take into account the variability inside each extraction methods (see the report protein analyses in the Supplementary Table S1). In this way, the total number of proteins identified was 39, 62, and 125 for trypsin shaving, biotinylation, and cell fractionation, respectively (Fig. 1). They were first subdivided into three broad categories according to their predicted localization: intracellular proteins or cytoproteins, cell envelope proteins, and exocellular proteins. Despite the precautions taken to avoid cell lysis and using surface-targeted extraction methods, a significant number of intracellular proteins were present in the three methods tested.
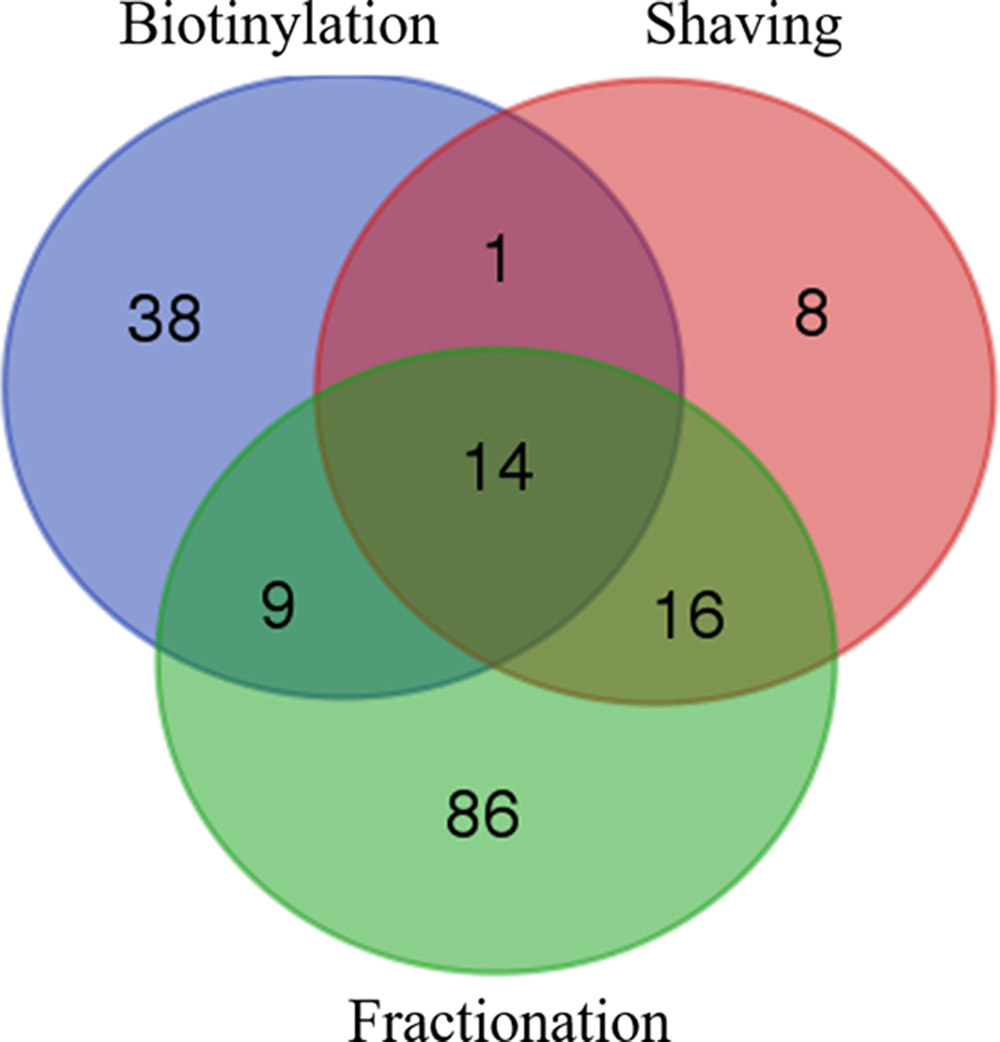
Venn-diagrams of total L. monocytogenes EGD-e proteins identified from the three extraction methods. For the cell fractionation method, the cell wall and membrane subfractions are pooled together.
Thus, 23, 30, and 100 cytoproteins were retrieved by trypsin shaving, biotinylation, and cell fractionation (considering both cell wall and membrane subfractions), corresponding to 59%, 48%, and 77% of total identified proteins in each of the fractions, respectively (Fig. 2). It is noteworthy that only one cytoprotein was identified in the membrane subfraction. The majority of the remaining proteins in the different samples were classified as cell envelope proteins (Renier et al., 2012). They represented 28% (11 proteins), 44% (27 proteins), and 19% (25 proteins) of the proteins extracted by trypsin shaving, biotinylation, and cell fractionation, respectively.

The number and relative percentages (in brackets) of L. monocytogenes EGD-e proteins identified with one or more unique peptides in at least two biological replicates by trypsin shaving, biotinylation, and cell fractionation considering cell wall and membrane subfractions, and according to subcellular localization predicted by a secretomics-based method (Renier et al., 2012).
In these last samples, 17 proteins were identified in the cell wall subfractions and 8 in the membrane subfractions, in which they represented an uneven proportion of 14% and 89% of the total proteins identified, respectively. This notable difference is related to the higher contamination of the cell wall subfractions by cytoplasmic proteins, contrary to the membrane subfractions but that contained a limited number of proteins. Finally, five proteins predicted to be extracellular were present in each of the samples from the three approaches, representing 4–13% of the total identified proteins.
Surface proteins identified by the three methods
To evaluate the efficiency and specificity of each method, all cell envelope proteins identified were distributed according to a Venn diagram (Fig. 3). Thanks to the biotinylation protocol, 16 proteins were specifically extracted, whereas the shaving and cell fractionation methods allow the specific extraction of 2 and 8 proteins, respectively. Seven proteins were extracted and identified by the three methods that represent a shared core of 18% of all surface proteins identified.
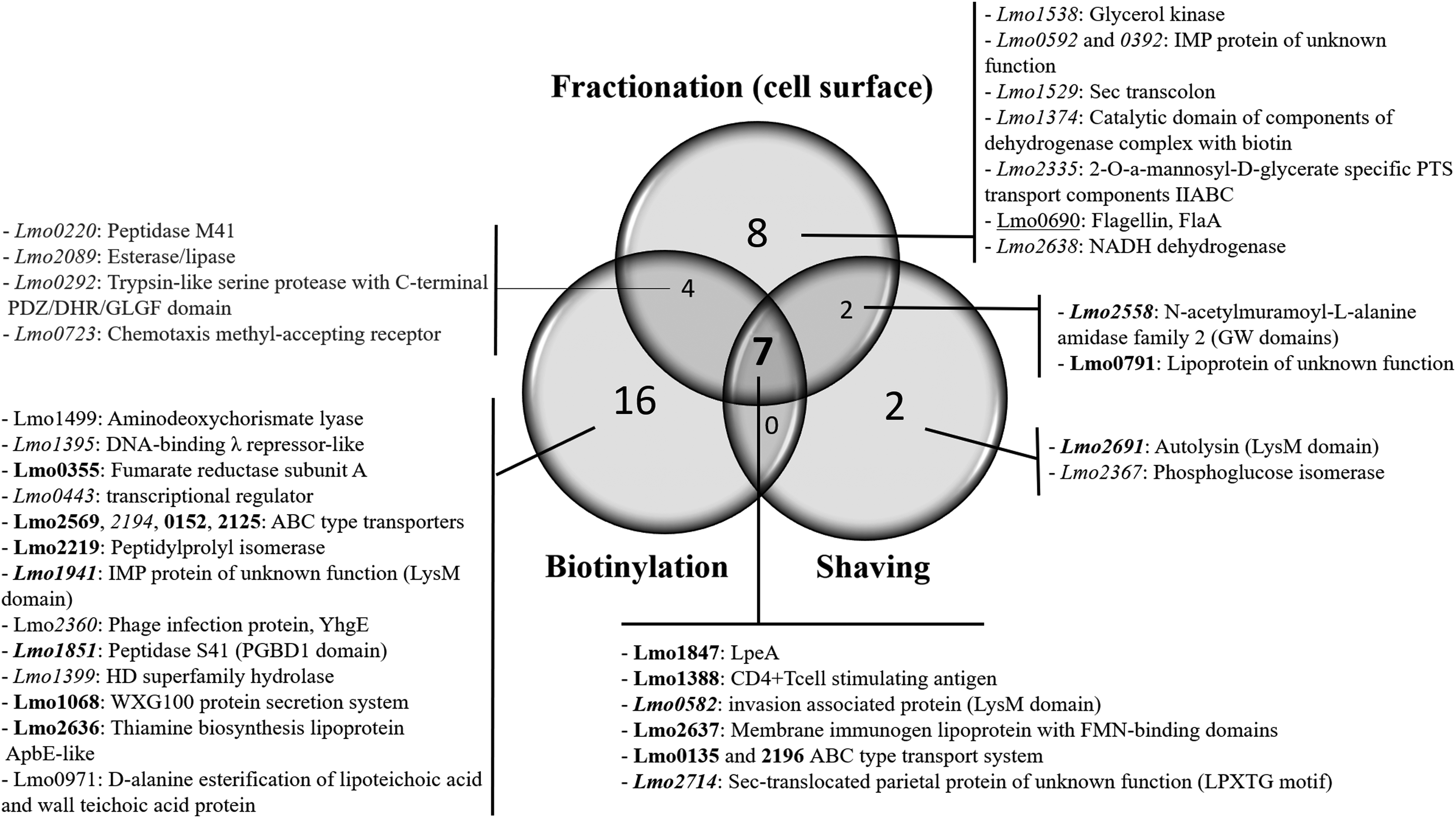
Venn diagram of cell surface proteins extracted with each method and their corresponding identification. Cell wall and membrane subfractions are pooled together in the fractionation method. In
Considering the cell fractionation method, three proteins were present in the membrane subfraction and four in the cell wall subfraction. The analysis of these 2 subfractions showed that among the 14 other surface proteins identified, only the protein Lmo0392 was identified in both the cell wall and membrane samples (data not shown). On the contrary, Lmo0690, Lmo2335, and Lmo2638 were specifically recovered in the membrane subfraction, whereas the lipoprotein of unknown function, Lmo0791, was found both in this subfraction and with the shaving method. The others proteins retrieved in the cell wall fraction were either specific (Lmo0592, Lmo1374, Lmo1529, and Lmo1538), common with the biotinylation (Lmo0220, Lmo0292, Lmo0723, and Lmo2089), or with the shaving (Lmo2558) methods (data not shown).
Predictive location of identified surface proteins
The accurate subcellular localization of the cell envelope proteins was defined using a secretomics-based prediction workflow compiling numerous bioinformatics tools (Renier et al., 2012) and taking into account the presence or absence of an SP, the secretion pathway, the binding motifs or domains to the cell wall or to the cytoplasmic membrane (Desvaux et al., 2006; Desvaux and Hébraud, 2009).
The biotinylation method enabled the extraction and identification of 2 exoproteins (Lmo0971 and Lmo1499), 12 lipoproteins, 9 cytoplasmic membrane-associated proteins (CMAPs), and 4 cell wall-associated proteins (CWAPs), whereas the shaving technique resulted in the identification of 6 lipoproteins, 1 CMAP and 4 CWAPs and the cell fractionation of 6 lipoproteins, 12 CMAPs and 3 CWAPs (Fig. 3). The lmo0690 gene product, namely the flagellin, which is a cell surface protein forming the filament of the bacterial flagella, was identified in the cell wall subfraction.
Discussion
The bacterial proteosurfaceome is relatively hydrophobic and can only be extracted from the cell membrane using detergents. Because of its variability and instability, its analysis present many hurdles including extraction, solubilization, purification, and analysis with in-gel (one- or two-dimensional gel electrophoresis) (Hebraud, 2014; Sun et al., 2011) or off-gel (shotgun proteomics) approaches (Cordwell, 2006).
Despite the challenge of working with surface proteins, their study remains an important research area, especially because of their role in the control of fundamental biochemical processes and their importance as pharmaceutical targets. Because proteins from the cell surface proteome of microorganisms are difficult to extract and identify using the conventional “in-gel” proteomics approach, particularly from Gram-positive bacteria, the efficacy of three extraction methods was evaluated on the pathogen L. monocytogenes.
Several methods, including the three evaluated in this study, have been previously assessed and compared, but on monoderm bacteria growing in planktonic mode (Dreisbach et al., 2010, 2011b; Hempel et al., 2011; Jessica et al., 2004; Tiong et al., 2015), and few have been performed on biofilms (Tiong et al., 2016). This growth mode in biofilms is the most widespread in the environment as noted earlier. The exploration of the proteosurfaceome of sessile bacteria is an additional challenge because of cell aggregation and the presence of extracellular compounds that form a matrix around the cells.
The three methods compared in this study are based on enzymatic shaving of the cell surface, biotin labeling of cell envelope proteins and cell fractionation, followed by an LC-MS/MS analysis of tryptic peptides.
Presence of intracellular proteins in cell surface proteome samples
Irrespective of the methodology used for cell surface proteome extraction and the measures used to avoid cell lysis, a significant number of intracellular proteins were present in the extracted samples, contaminating the proteosurfaceome.
In the case of enzymatic shaving, this contamination may be because of the lysis of some bacteria within the biofilm or the alteration of the cell envelope during the preparation of the proteosurfaceome samples. The frailty or robustness of the cell envelope to the surface extraction process may differ depending on whether addressing a monoderm or diderm ( = Gram-negative) and on the genus and bacterial species.
Consequently, some methodological parameters must be adapted to the microorganism studied, such as temperature, incubation time, buffer for enzymatic digestion (Dreisbach et al., 2010), quantity of enzyme, and centrifugation speed to recover cells. These parameters influence both the efficiency to extract surfaceome peptides and the degree of contamination by intracellular proteins.
The surface proteins identified in this study on L. monocytogenes biofilms were consistent with those reported in the literature on planktonic cells (Tjalsma et al., 2008; He and De Buck, 2010; Olaya-Abril et al., 2012), despite the presence of a significant proportion of cytoplasmic proteins.
For the biotinylation method, L. monocytogenes intact cells were treated with Sulfo-NHS-SS-Biotin, a marker molecule that is supposed to be membrane impermeable. In fact, this technique, initially implemented on eukaryotic cells has been applied on diderm bacteria by using different biotinylation reagents whose Sulfo-NHS-SS-Biotin seemed the most suitable, even if some periplasmic, cytoplasmic, and inner membrane proteins were identified (Hempel et al., 2011; Monteiro et al., 2018).
With the cell fractionation methods, the washing steps to remove potential contaminating intracellular proteins were efficient for the membrane subfractions but to a lesser extent for the cell wall subfractions. Intracellular proteins seem to be trapped or to have an affinity for components of the cell envelope and thus to be carried along them in the subsequent steps of the extraction process.
In addition, among the proteins considered as contaminants, certain proteins may have a double localization with a different function than known at the cytoplasmic level. Some of these proteins can be moonlighting proteins that is exhibiting an additional function when localized at the bacterial cell surface. Similarly, some secreted exoproteins may be in transit from the cytoplasm and be retrieved in the proteosurfaceome. We must also consider that the subcellular localization of proteins predicted as cytoplasmic maybe incorrect or incomplete because they potentially harbor unknown motifs for cell-envelope attachment or their secretory pathway could not be identified.
Despite these drawbacks, the optimization of each of these methods should not be at the expense of their efficiency in extracting proteins or peptides from the bacterial cell surface and the presence of cytosolic proteins is not critical as it does not interfere with their identification by mass spectrometry.
Comparison of the three methods
Among the various strategies for exploring the proteosurfaceome, the enzymatic shaving method has been applied in recent years, mainly on monoderm bacteria (Solis and Cordwell, 2011), with contrasting results depending on the bacterial species. Shaving method has been previously used to explore the cell surface proteome of a wide variety of microorganisms (Olaya-Abril et al., 2014), among them Gram-positive pathogens such as L. monocytogenes (Tiong et al., 2015), Streptococcus spp. (Pribyl et al., 2014; Rodriguez-Ortega et al., 2006; Severin et al., 2007), Staphylococcus aureus (Dreisback et al., 2010, 2011a, 2011b; Monteiro et al., 2015; Solis et al., 2010; Pribyl et al., 2014; Tiong et al., 2015; Ventura et al., 2010), and other monoderm bacteria such as Bacillus subtilis (Tjalsma et al., 2008) or the dairy starter Lactococcus lactis (Meyrand et al., 2013).
This technique has been successfully used to explore L. monocytogenes proteosurfaceome with 19 surface proteins of a total of 174 proteins identified by Zhang et al. (2013) or 18 proteins of 155 proteins identified by Tiong et al. (2015). In this study, 11 surface proteins were identified from a total of 39 proteins, with 6 lipoproteins, 1 membrane and 4 cell wall proteins. In comparison, our samples had less contamination by intracellular proteins but fewer surface proteins were characterized. This can be a consequence of the biofilm mode of growth, reflecting a more difficult access of the trypsin to the cell surface because of the presence of extra polymeric substances.
By using another protocol to extract proteins from the cell envelope, called UB-Ghost, and implementing a hypotonic extraction buffer and then an 8 M urea buffer before trypsin digestion, Tiong et al. (2016) showed similar results. Indeed, they identified 14 and 109 surface proteins from sessile and planktonic cells, representing 2.3% and 17.6% of the total proteins identified, respectively (Tiong et al., 2016).
Concerning Streptococcus spp., shaving seemed more efficient than for L. monocytogenes because it allowed the identification of 68 and 58 surface-localized proteins in Streptococcus group A (Rodriguez-Ortega et al., 2006) and Streptococcus pyogenes (Severin et al., 2007), respectively. However, by using immobilized trypsin on agarose beads, Pribyl et al. (2014) identified only six surface proteins in Streptococcus pneumoniae (four lipoproteins and two Leu-Pro-any-Thr-Gly motif [LPXTG]-anchored proteins). All these proteins were also identified by the biotinylation method; this approach was also more effective than shaving with 39 surface proteins identified (29 lipoproteins and 10 LPXTG-anchored proteins) (Pribyl et al., 2014).
The biotin-labeling method of bacterial cell envelope protein has been mainly performed on diderm bacteria and few attempts on monoderm bacteria have been reported to date. The biotinylation approach was also applied on the vancomycin-resistant Enterococcus faecium E155, allowing the extraction of 43 proteins, among which 27 proteins were with solely extracytoplasmic localization (Romero-Saavedra et al., 2014). The shaving method on the same bacterium was more efficient with 39 extracytoplasmic proteins identified, although the original sample was severely contaminated by cytoplasmic proteins (390 proteins identified in total).
The cell surface proteome of the opportunistic pathogen Staphylococcus aureus was one of the first explored by a range of different approaches. For example, Solis et al. (2010) evaluated the cell shaving method by using trypsin and proteinase K. The overlap between the two enzymes resulted in the identification of 10 surface-exposed proteins, whereas 12 and 16 were specific to trypsin and proteinase K, respectively, reflecting both their specific cleavage and their complementarity. In addition, the proteins were classified as lipoproteins and proteins with a Lysin motif (LysM) (noncovalent) or LPXTG (covalent) motifs bounding to peptidoglycans, demonstrating the efficacy of the enzymatic method.
Even more conclusive results from trypsin shaving were reported with the identification of 72 (Dreisbach et al., 2011b) and 101 surface-associated proteins (Hempel et al., 2011), about half of which were transmembrane proteins, the other half belonging to the other categories listed previously. By comparison, the biotinylation implemented in each of these two studies allowed to identify 49 and 110 cell-surface proteins, respectively. In the study of Hempel et al. (2011), about two-thirds of the total proteins identified, and predicted as surface associated, were common to both methods (i.e., 66/101 and 110 for shaving and biotinylation, respectively) (Hempel et al., 2011).
Noticeably, by combining the 2 methods, almost all lipoproteins were identified by biotinylation (39 of 40), whereas all the 15 sortase substrates, proteins covalently linked to the cell wall, were identified by trypsin shaving. We observed similar results for L. monocytogenes, 13 lipoproteins were identified in total using both methods with 12 found with biotinylation, 7 of which specific to this method. On the contrary, the two approaches did not reveal any specificity at the level of sortase substrates and/or cell wall proteins. It should also be noted that the conventional cell fractionation approach was the most effective in identifying membrane proteins with 11 of the 17 proteins obtained by the three methods we compared, including 7 specific proteins.
Focus on cell surface proteins identified
The cell envelope of Gram-positive bacteria ( = monoderm) is primarily composed of a single biological membrane and a cell wall of peptidoglycan (Shockman and Barrett, 1983). To ensure their functions, surface proteins are both targeted to the cytoplasmic membrane and the cell wall (Desvaux et al., 2006). Membrane-associated proteins include membrane-integrated proteins, peripheral membrane proteins, and lipoproteins. Anchoring to the cell wall is allowed by two primary mechanisms: (i) cell wall sorting and (ii) targeting (Navarre and Schneewind, 1999). Cell wall sorting is the covalent attachment of surface proteins to the peptidoglycan through a C-terminal sorting signal that contains a consensus LPXTG sequence.
Cell wall targeting involves the noncovalent attachment of proteins to the cell surface through specific binding domains (Desvaux et al., 2006, 2018). Six cell wall-associated proteins were recovered with the combination of the three extraction methods, with two containing a LysM motif (Buist et al., 2008). This motif is found in six L. monocytogenes proteins (Bierne and Cossart, 2007), including the invasion-associated protein Iap (Lmo0582). This protein, also called CwhA, has been characterized as an autolysin with a relevant role in infection (Dussurget et al., 2004; Lenz et al., 2003). Two other proteins containing noncovalent cell wall-binding domain were identified, the peptidase S41 (Lmo1851) with a PGBD1 domain and Lmo2558 with a GW module.
A protein of unknown function containing an LPXTG motif for covalent binding to peptidoglycan (Lmo2714) was also retrieved. This family of LPXTG proteins, firmly anchored to the cell wall, comprises 41 proteins in L. monocytogenes (Cabanes et al., 2002). Considering that only few proteins with LPXTG motif were recovered in our study, the three methods tested do not seem efficient to monitor them. To access these proteins strongly associated with peptidoglycans, a method based on boiling cell envelopes of L. monocytogenes in the presence of SDS was previously shown more effective and allowed to identify 13 LPXTG-containing proteins (Graciela et al., 2005).
Based on their characteristic signal sequences and conserved lipobox motif in the C-domain (Tjalsma et al., 2000), 68 of 133 surface proteins were classified as lipoproteins in L. monocytogenes EGD-e strain (Glaser et al., 2001), and 26 were later confirmed in situ (Baumgartner et al., 2007). Almost half of these lipoproteins are presumed to act as substrate-binding components of ATP-binding cassette (ABC) transporter systems (Bierne and Cossart, 2007), performing the equivalent functions of periplasmic solute-binding proteins in Gram-negative bacteria (Tam and Saier, 1993). Despite their predominance as surface proteins, very few lipoproteins have been biochemically characterized like LpeA (Lmo1847), which belongs to the LraI family of manganese-importing ABC transporter components (Novak et al., 1998) and TscA (Lmo1388), a CD4+ T cell-stimulating antigen.
Concerning the CMAPs, 17 were identified, mainly by the biotinylation and cell fractionation methods. The CMAPs are anchored within the lipid bilayer and systematically exhibit at least one α-helical TMD and sometimes multimembrane-spanning domain as is the case for Lmo2335, Lmo2360, or Lmo2638 (Desvaux and Hébraud, 2009; Renier et al., 2012). Thus, the CMAPs are classified according to the TMD topology (Desvaux and Hébraud, 2008) but irrespective of their topology, these hydrophobic TMD domains make their extraction challenging by classical proteomic approaches.
The cell fractionation allowed the identification of the flagellin in the cell wall fraction. This cell surface protein, exported through the flagella export apparatus, is the constitutive subunit of the flagella filament. FlaA exhibits also a peptidoglycan-hydrolyzing activity that might play a role during flagella assembly (Popowska, 2004; Popowska and Markiewicz, 2004). Indeed, some flagellar components are assembled on the bacterial cell surface like for the filament proteins FlaA and FliD (Desvaux and Hébraud, 2008), which could explain its presence in the cell wall subfraction.
Overall, the three methods implemented in our experimental conditions allowed to extract and to identify 39 proteins of the 133 L. monocytogenes surface predicted proteins (29%). A relatively low recovery rate (18%) was found between the different methods with only seven surface proteins in common, highlighting the complementarity of these approaches.
Conclusion
The cell surface proteome comprises proteins usually difficult to extract and is subject to several factors of variation such as mode (planktonic or sessile), phase, and conditions (e.g., medium, temperature and pH) of growth, including differences between monoderm and diderm bacteria. In this study, we used three different extracting methods for exploring the proteosurfaceome of L. monocytogenes. The three methods were applied on bacterial cells growing in a biofilm, which is the most widespread growth mode in natural and agri-food environments of this pathogen.
The proteosurfaceome, because of its importance for adhesion, biofilm formation, and bacterial persistence in natural and in food-processing environments, requires particular attention. Despite the lack of abundant extra polymeric substances in L. monocytogenes biofilms, accessing the cell surface proteome of these sessile and aggregated bacteria for their proteomic analysis is extremely challenging and presents more difficulties than for planktonic cells.
Our results demonstrated that a multiapproach strategy, taking advantage of the complementary of several methods, is needed to fully explore the cell surface proteome of L. monocytogenes in biofilm. Other additional approaches have been shown to be effective on planktonic bacteria and could be used to further expand the analysis of the biofilm proteosurfaceome. Among them, the Ghost urea method has been already used on adhered cells (Tiong et al., 2016). Moreover, the method of using cell wall extracts boiled with SDS before their trypsin digestion (Graciela et al., 2005) could allow to target more specifically the LPXTG proteins.
In conclusion, these different methods for deciphering the proteosurfaceome often require modifications and improvements according to the bacterial species, and their selection depends on whether a profiling or a targeted analysis is required. These new findings collectively inform future discovery and translational proteomics for clinical, environmental health, and industrial applications.
Footnotes
Acknowledgments
We thank Mathilde Cohadon for her support in biotinylation analysis and Ingrid Chafsey for technical assistance in providing the biotinylation protocol. J.E. was granted from the Agence Nationale de la Recherche (ANR) (France) as part of an ANR-12-ALID-0005 EcoSec contract. T.S. is a Marie Sklodowska-Curie, PhD Research Fellow granted by ITN ETN List-MAPS.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.