
Abstract
Abstract
The human epigenome plays a key role in determining cellular identity and eventually function. Drug discovery undertakings have focused mainly on the role of genomics in carcinogenesis, with the focus turning to the epigenome recently. Drugs targeting DNA and histone modifications are under development with some such as 5-azacytidine, decitabine, vorinostat, and panobinostat already approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA). This expert review offers a critical analysis of the epigenomics-guided drug discovery and development and the opportunities and challenges for the next decade. Importantly, the coupling of epigenetic editing techniques, such as clustered regularly interspersed short palindromic repeat (CRISPR)-CRISPR-associated protein-9 (Cas9) and APOBEC-coupled epigenetic sequencing (ACE-seq) with epigenetic drug screens, will allow the identification of small-molecule inhibitors or drugs able to reverse epigenetic changes responsible for many diseases. In addition, concrete and sustainable innovation in cancer treatment ought to integrate epigenome targeting drugs with classic therapies such as chemotherapy and immunotherapy.
Introduction
All somatic cells contain the same genetic material, but display varied cellular phenotypes. Phenotypic variability is determined not only by genome but also the epigenome. The interaction of the genome and the epigenome is universal in all multicellular organisms and necessary for developmental biology and function.
Epigenetics was coined by Waddington in 1942 as the alteration of the phenotype with no obvious alteration of the genotype, resulting in developmental changes that are not accompanied by genetic changes (Allis and Jenuwein, 2016; Waddington, 1942). Most epigenetic alterations resulting in diseases involve mutations in noncoding regions of the DNA that can cause epigenetic changes, loss of function mutation that stops the function of an epigenetic component, and inherited aberrant epigenetic marks. Covalent modifications of the DNA, RNA, and histones are primarily what make up the epigenome (see Box 1 for definitions). Enzymes responsible for the application of the covalent modifications are called “writers” (Table 1).
Writers, Readers and Erasers Involved in Covalent Modifications of Chromatin
Cellular reprogramming: Inducing of pluripotency in a differentiated cell
Driver gene: An important gene whose mutations can cause its activation or deactivation of its expression leading to malignant transformation of a cell
Epigenetics: Alterations of the phenotype with no obvious alterations of the genotype
Erasers: Enzymes responsible for removal of histone modifications from chromatin
Hypermethylation: Elevated methylation of DNA or histones
Hypomethylation: Reduced methylation of DNA or histones
Immune checkpoint inhibitors: These are inhibitors of immune checkpoint proteins responsible for keeping immune responses in check and thus prevent immune cells from attacking cancer cells
Plasticity: The ability to reverse epigenetic modifications of DNA and histones
Readers: Proteins that bind to chromatin through recognition of specific histone modifications
Writers: Enzymes that add histone modifications to the chromatin
Proteins that recognize the covalent modifications are called “readers” and these function to remodel certain genomic regions that ultimately influence the expression of many genes (Table 1). Enzymes that are responsible for the removal of covalent modifications are called “erasers” and gives rise to the flexibility of the epigenome (Table 1). Depending on the prevailing situation, epigenetic modifications can have different functions.
Many international initiatives, aimed at mapping the human epigenome, have been carried out or are underway and include the International Human Epigenome Consortium, BLUEPRINT, ENCODE, International Cancer Genome Consortium, and The Cancer Genome Atlas Research Network (The Cancer Genome Atlas Research Network, 2017; Fernandez et al., 2016; Laperle et al., 2018). These initiatives have been at the forefront of mapping the human epigenome in different tissues and pathological conditions (2017; Shukla, 2017).
This expert review offers a critical analysis of the epigenomics-guided drug discovery and development and the opportunities and challenges for the next decade. In addition, concrete and sustainable innovation in cancer treatment ought to integrate epigenome targeting drugs with classic therapies such as chemotherapy and immunotherapy.
Literature Search
A literature search of the PubMed and Google Scholar was performed for pertinent published articles in the English language until December 2018. The search keywords included epigenetics, cellular reprogramming, DNA methylation, histone modification, cancer, drug development, inhibitors, small molecules, tumor heterogeneity, cancer modeling, personalized medicine, and drug development. Relevant full articles were retrieved for the review (Fig. 1).
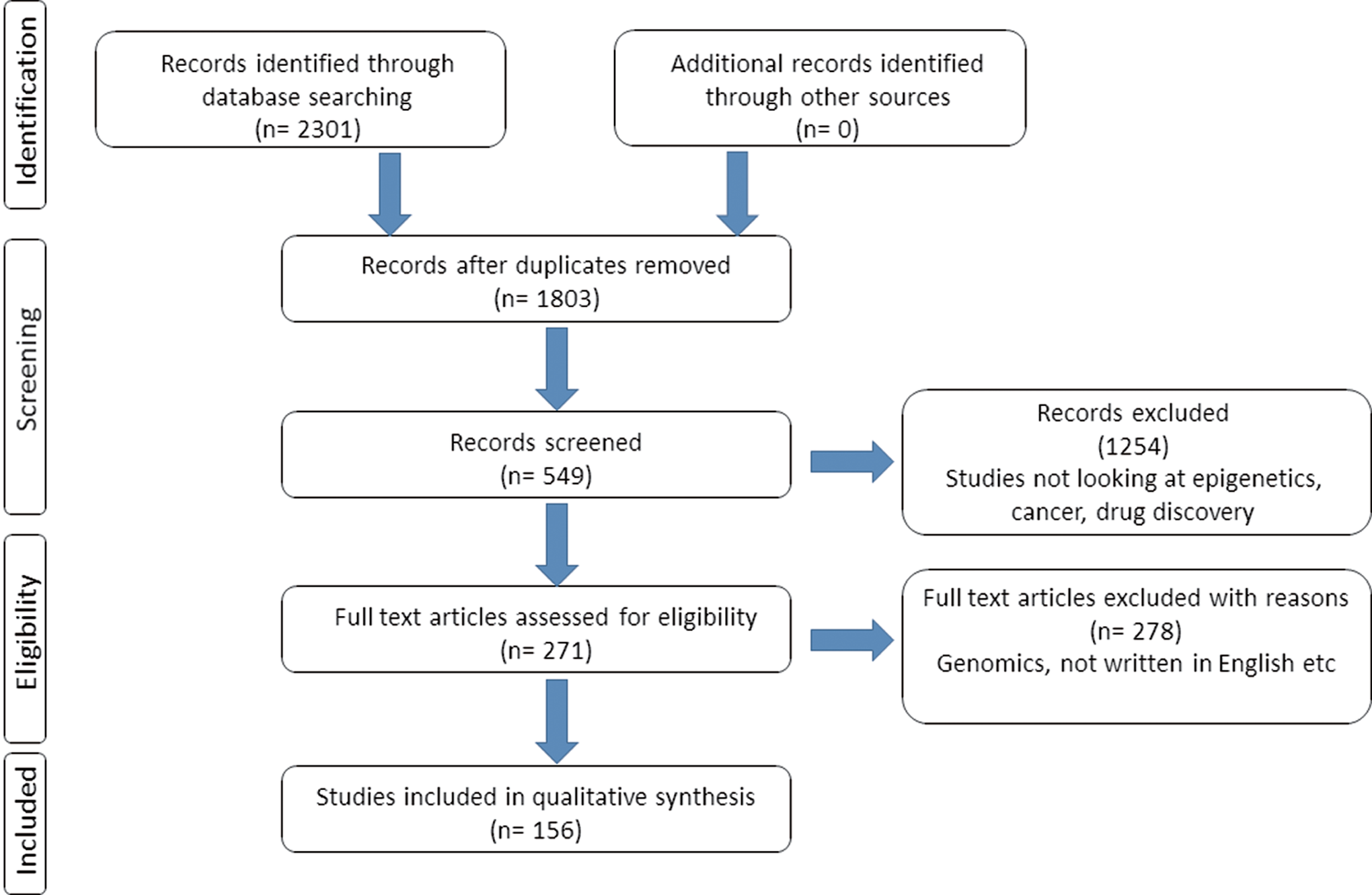
Search strategy and selection criteria of the literature used in this review.
A total of 2301 articles were consulted after search; exclusion criteria were performed based on article relevance to the expert review. Subsequently, 156 articles that gave required information were included in this review. The articles selected focused on research, clinical trials, and reports describing the above words. This thorough search allowed the writing of a comprehensive review on epigenetic drug development and its future. Many authors have contributed significantly to the study of epigenetics, drug discovery, and development over decades. While not all studies could be cited here as a focused expert review, the leading concepts and example articles are distilled and discussed throughout the article.
Epigenetic Alterations Observed in Tumors
Next-generation genome sequencing of tissues and tumors has shown that many epigenetic mechanisms are altered in tumors, with many enzymes containing many mutations (Kretzmer et al., 2015; Weinstein et al., 2013). These mutations cause different cancer cell characteristics ranging from uncontrolled cellular growth, cancer cell heterogeneity, drug resistance, and metastasis (Arrowsmith et al., 2012; Pfister and Ashworth, 2017). In cancers, both genomic and epigenomic aberrations continue to interact, presenting a conundrum to scientists in terms of understanding the cause of cancer, heterogeneity, and therapy resistance (Jones et al., 2016; You and Jones, 2012).
It has been observed that mutations in genes encoding chromatin modifying enzymes are common in both childhood and adult cancers, with aberrant methylation of DNA (Bayliss et al., 2016; Mack et al., 2014). For example, myeloid malignancies typically display mutations in isocitrate dehydrogenase 1 (IDH1) and IDH2 genes and TET2 gene, interfering with the activity of demethylases (Figueroa et al., 2010; Lu et al., 2012; Yamazaki et al., 2015).
Recent data show that many genes involved in the regulation of the chromatin are mutated in cancers and cause alterations of the epigenome (Falahi et al., 2015; Jones et al., 2016; Rasmussen et al., 2015). Compared to normal cells, tumor cells display aberrant DNA methylation patterns, displaying decreased methylation on DNA or histones throughout the genome, while showing increased methylation at gene promoters (Baylin and Herman, 2000; Jones and Laird, 1999).
Epigenetic mutations also result in abnormal histone modifications and abnormal chromatin structures. Decreased methylation of the genome causes genomic instability and can activate oncogenes (Gaudet et al., 2003; Yang et al., 2003). Increased methylation at gene promoters causes gene silencing and can affect tumor suppressor genes (Baylin, 2005; Rhee et al., 2002). Causes of the abnormal DNA methylation patterns include diet, physical exercise, and age (Fernandes et al., 2017; Horsburgh et al., 2015).
Therapeutic exploitation of epigenetic alterations is appealing due to the fact that some of the mutations to epigenetic genes are known to be drivers of cancer development and are prevalent in many cancers (Pfister and Ashworth, 2017). Most importantly, one epigenetic mutation might affect cancer-related processes, its targeting resulting in many beneficial effects. Due to their reversibility, epigenetic modifications might be used to reactivate silenced tumor suppressor genes.
Epigenetic Disruptions Are Key to Carcinogenesis
The traditional view of cancer as a collection of diseases caused by genetic aberration accumulation leading to neoplasia is now known not to be entirely true (Hanahan and Weinberg, 2011; Kaneda et al., 2014). Successive accumulation of disruptions to growth control mechanisms for important cellular processes such as proliferation, angiogenesis, cell death, and migration results in malignant cells (Choi and Lee, 2013; Shen and Laird, 2013). The resultant malignant properties are then stably inherited by subsequent clones. These malignant properties are brought about by different means from copy number variations, deletions, insertions, recombination, and mutations (Baylin and Jones, 2011b; Garraway and Lander, 2013).
Genetically identical cells may have different phenotypes as a result of epigenetic changes influencing processes such as transcription. The epigenetic mechanisms implicated in such scenarios include DNA modification, methylation of CpG cytosine-5, hydroxylation and formylation, histone modifications, noncoding RNAs, and carboxylation (Jones, 2012; You and Jones, 2012). By acting in unison, epigenetic mechanisms collaborate to drive cellular malignant transformation. These epigenetic signatures also change depending on microenvironmental conditions and niches.
Since epigenetic alterations are reversible, factors such as age, environment, and mutations can change the state and function of the epigenome (Fig. 2). The cooperation between mutations and epigenetic changes combine to transform normal cells and to provide the transformed cells with certain advantages over normal cells (Boumahdi and Blanpain, 2016; Kaufman et al., 2016). Thus, the epigenomes of cells undergoing transformation play a huge role in the phenotype displayed by the eventual tumor cells (Oakes et al., 2016).
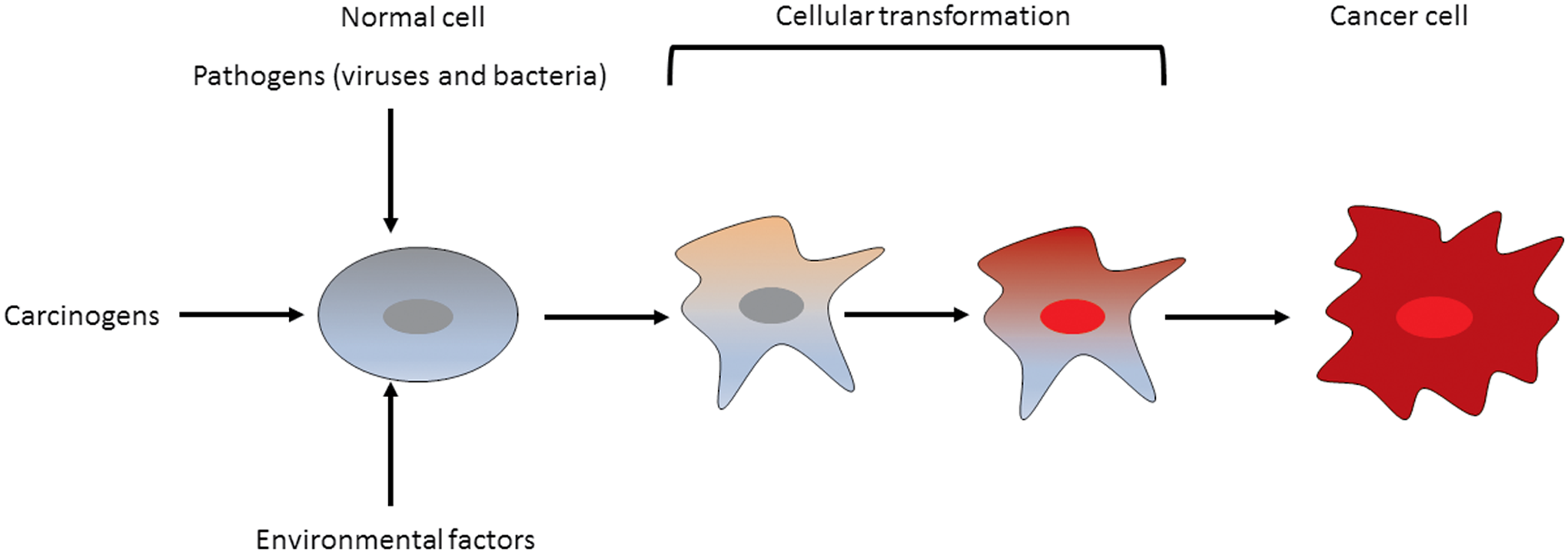
Both genetic and epigenetic changes can occur in a cell leading to its transformation. These changes are caused by intrinsic and extrinsic factors such as mutations and pathogens, respectively. The synergistic effect of mutations and epigenetic changes can provide a growth advantage and a fertile ground for cellular evolution leading to growth of a tumor.
It is now accepted by most geneticists that alterations of the epigenome is as important to carcinogenesis as genetic mutations (Chen et al., 2014; Sui et al., 2015). In fact, epigenetic alterations might precede carcinogenesis (Baylin and Jones, 2011a). The deamination of 5-methylcytosine (5mC) to thymine is known to induce many mutations in cells, supporting the idea of epigenetic changes giving rise to changes in the genome (Herceg et al., 2018; Joubert et al., 2016). In addition, nutrition-, environment-, and age-related changes to the epigenome can be drivers of the tumorigenic process (Dzobo et al., 2018a; Vogelsang et al., 2014). For example, it has been suggested that Vitamin C can contribute to DNA hypermethylation and alter gene expression (Blaschke et al., 2013; Dickson et al., 2013).
Furthermore, inflammation can cause cellular stress and has been suggested to cause epigenetic changes in chromatin structure and methylation levels of DNA (Yara et al., 2013). Any change to the epigenome and genome is likely to influence gene expression and can give rise to transformed cells. Thus, targeting both epigenetic and genetic changes to restore normalcy in the cell represents some of the proposed strategies to treat cancer.
A brief discussion of the epigenetic changes that influence the expression of several important genes leading to cancer development is given below.
DNA Methylation
DNA methylation has been studied in cancer as the epigenetic mark resistant to sample preparations (Houshdaran et al., 2010; Laird, 2010). In mammals, DNA methylation is known to occur mostly at the so-called CpG islands, resulting in gene silencing (Jeong et al., 2009; Jones and Liang, 2009). CpG islands represent almost 50% of promoters and most of them are unmethylated. DNA methyltransferases (DNMTs) catalyze the transfer of methyl groups from S-adenosyl-
It has been shown that DNMT1 is responsible for maintaining methylation patterns during cell division (Sharma et al., 2010a, 2010b, 2010c). The patterns of DNA methylation are influenced by the sequence of DNA and by germline variation (Gertz et al., 2011; Tang et al., 2016). The germline contains unmethylated CpG islands and this is maintained by guanine-cytosine strand asymmetry. Mutation in DNMTs such as DNMT1, DNMT3A, and DNMT3B has been described in colorectal cancer, myelodysplastic syndromes (MDS), and chromosomal instability (Kanai et al., 2003; Shirahata-Adachi et al., 2017; Wang et al., 2017a; Zaker et al., 2017). Mutations in DNMT3A are also known to be involved in acute myeloid leukemia (AML) disease progression (Russler-Germain et al., 2014; Shivarov et al., 2013; Yang et al., 2015).
Proteins such as methyl-binding domain (MBD) proteins have been implicated in mediating transcriptional repression when they bind to CpG islands (Du et al., 2015; Lang et al., 2015). These include MBD1, MBD2, and MBD 4. Genetic aberration in MBD proteins such as MBD 1 and MBD2 is increased in cancers such as breast and lung cancer (Baubec et al., 2013; Guerrero-Preston et al., 2014). The demethylation of DNA is known to occur through a passive or an active process. In the passive process, DNA methylation is impaired during DNA replication, with the new DNA strand having no methylation (Kagiwada et al., 2013; Ohno et al., 2013). On the other hand, the active DNA demethylation process depends on enzymes for the hydroxylation, oxidation, and deamination of 5mC and does not have to occur during DNA replication (Kawasaki et al., 2014; Lee et al., 2014; Wu et al., 2014).
The so-called “erasers” of DNA methylation include activation-induced cytidine deaminase (AID) and ten-eleven translocation proteins (TETs) (De Carvalho et al., 2010; Kelly et al., 2010). The demethylation of DNA through the active process is known to be a sequence of events starting with the hydroxylation of 5mC by TET proteins followed by the deamination by AID/APOBEC protein, or sometimes the carboxylation and entry into the subsequent base excision repair pathway (Bhutani et al., 2011). Of the TET family proteins, TET2 mutations are found in myeloproliferative neoplasms.
Studies on MLH1 and CDKN2A genes in a colon cancer cell line, HCT116, show that one allele of each gene is mutated, while the other allele is DNA methylated, and thus silenced (You and Jones, 2012). Processes such as DNA repair and cell cycle regulation suffer due to no expression of these two genes. This is a clear example of genetic-epigenetic cooperation in causing mutations that will ultimately cause cancer.
The WNT signaling pathway in normal cells is antagonized by the secreted frizzled related proteins (sFRPs). Overexpression of the WNT signaling pathways results from the silencing of the sRFPs. Overexpression of the WNT signaling results in increased and uncontrollable cellular proliferation. Such cells have survival advantages over normal cells, hence will continue accumulating genetic aberrations leading to cancer. Data from next-generation sequencing and other global studies have shown that certain cancers have mutations such as p53 as the predominant ones.
Histone Modifications
The basic building unit of chromatin are the nucleosomes and they are made up of DNA wrapped in histones (Luger et al., 2012). It is known that histones can change the structure of chromatin (Gonzales-Cope et al., 2016). Some of the enzymes involved in the histone modifications include histone deacetylases (HDACs) and histone acetyltransferases (HATs). These enzymes do remove and add acetyl groups to the histones. The enzymes that add and remove methyl groups are called histone methyltransferases and histone demethylases.
During carcinogenesis, there are wide changes that occur and these affect the recruitment of transcription factors, and thus add to the aberrant gene expression observed (Sharma et al., 2016). HATs can be grouped as p300/CBP, PCAF, and MYST. HDACs are notorious for the silencing of tumor suppressor genes (You and Jones, 2012). This is why HDAC inhibitors have been used as anticancer drugs (Negmeldin et al., 2017; Poli et al., 2017). Modifiers of the histones also work as complexes, the classical example being the Trithorax and Polycomb complexes (Mills, 2010). These complexes antagonize each other in controlling developmental genes and have been implicated in cancer development (Mills, 2010). Both complexes lay down histone marks to either repress or activate transcription of several genes.
In cancers such as colon, gastric, and epithelial cancers, mutations in HDAC2 have been associated with resistance to HDAC inhibitors (Smith et al., 2010). On the flip side, HDACs have been shown to act as tumor suppressors through mechanisms, including genome stabilization (Bhaskara, 2015; You and Jones, 2012). The presence of chromatin binding proteins such as bromodomain (BRD) proteins can also affect the process of tumorigenesis. Aggressive midline carcinoma develops when BRD proteins bind to nuclear protein in testis (NUT) (Bragelmann et al., 2017; Wang et al., 2017b).
Aberrant expression of BRD proteins has been reported in many cancers, making these proteins potential targets for therapy (Grayson et al., 2014; Sholl et al., 2015). The self-renewal of cancer stem cells has been shown to be regulated by BMI-1 and its overexpression has been observed in cancers, including prostate (Kumar et al., 2017; Li et al., 2017a). Cancer stem cells have been shown to be present in many cancers, including colon, prostate, and breast cancers (Dzobo et al., 2016). BMI-1 is a component of Polycomb repressive complex 1 (PRC1) and is also important in the differentiation of T cells (Miyazaki et al., 2008; Raaphorst et al., 2001).
Epigenetic Drug Development
Many epigenetic drugs are being developed or already approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA). In vitro analysis of many small molecules is followed by phenotypic assays, genomic, epigenomic, transcriptomic, and proteomic analyses (Fig. 3). Potential inhibitors are then investigated using disease models such as 3D organoids and mice models of cancer. Candidate molecules are then tested under clinical settings.
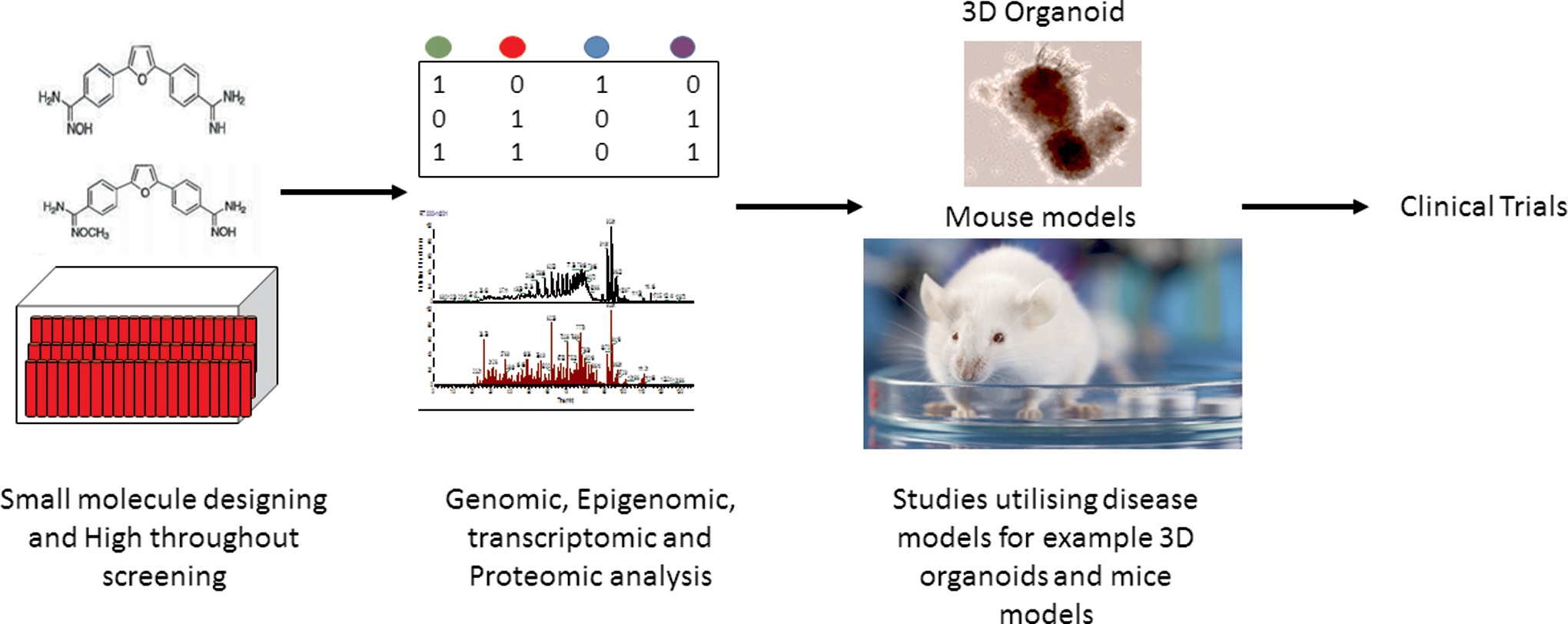
Schematic depiction of the flow of drug design and analysis. In vitro analysis of many small molecules is followed by phenotypic assays, genomic, epigenomic, transcriptomic, and proteomic analyses. Potential inhibitors are then investigated using disease models such as 3D organoids and mice models of cancer. Candidate molecules are then tested under clinical settings.
DNMT inhibitors
Many cancers, including lung, breast, colorectal, and gastric cancers, display increased methylation levels of tumor suppressor gene promoters and overexpression of DNMTs (Liu et al., 2017). Inhibitors of DNA methylation can therefore lead to reduced promoter hypermethylation and can activate silenced tumor suppressor genes, including p16 and CDKN2A (Pfister and Ashworth, 2017; Yoo and Jones, 2006). DNMT inhibitors can be nucleoside analogues and non-nucleoside analogues. Nucleoside analogues are incorporated into DNA and trap DNMTs onto DNA, while non-nucleoside analogues bind to the catalytic region of DNMTs (Brueckner et al., 2005). Well-known DNMT inhibitors and nucleoside analogues are 5-azacytidine (Celgene Corporation), decitabine (Otsuka Pharmaceutical), and guadecitabine (Astex Pharmaceuticals) (Fig. 4) (Sorm and Vesely, 1964).

DNMT inhibitors decitabine, 5-azacytidine, and guadecitabine. DNMT, DNA methyltransferases.
These inhibitors remove hypermethylation of tumor suppressor genes. While 5-azacytidine and decitabine have been approved by both the FDA and EMA for various cancers, guadecitabine is still at different phases of clinical trials for different malignancies (Jones et al., 2016). For example, guadecitabine is under phase II clinical trial for use in patients with higher risk MDS and AML (Trial Number: NCT02131597). It is important to note that guadecitabine is being trialled in combination with chemotherapy and immune checkpoint inhibitors for solid tumors (Ribich et al., 2017). Other DNMT inhibitors, including natural compounds such as genistein and epigallocatechin gallate, are undergoing different stages of development and clinical trials (Gnyszka et al., 2013).
One major worry regarding the use of DNMT inhibitors has been their toxicity. By causing reduced DNA methylation, DNMT inhibitors can randomly reactivate many genes with deleterious effects to cells (Gnyszka et al., 2013; Yoo and Jones, 2006). While it is good that DNMT inhibitors can reprogram cancer cells to normal cells, our group has shown that 5-azacytidine can also reprogram mesenchymal stem cells to “tumor-associated fibroblasts” (Senthebane et al., 2017). Currently, DNMT inhibitors are administered at low doses and being used to sensitize cancer cells to conventional therapy such as chemotherapy (Juergens et al., 2011).
HDAC inhibitors
The acetylation of histones leads to increased chromatin accessibility to transcription factors. HATs and HDACs are the enzymes responsible for this modification and the overexpression of HDACs is observed in many cancers, including cervical, colorectal, and prostate cancers (Li and Seto, 2016). By inhibiting histone deacetylation and therefore increasing histone acetylation, HDAC inhibitors can reduce oncogene transcription and reactivate silenced tumor suppressor genes such as p21 (Rocchi et al., 2005). HDAC inhibitors can also modulate the activities of p53 and NF-kB transcription factors (Chen and Greene, 2003; Gu and Roeder, 1997). Ultimately, HDAC inhibitors are known to kill cancer cells through DNA damage, cell cycle arrest, induction of apoptosis, and ROS generation (Cappellacci et al., 2018).
Well-known FDA-approved HDAC inhibitors include belinostat (Spectrum Pharmaceuticals), vorinostat (Merck and Co.), and romidepsin (Celgene) (Eckschlager et al., 2017; Lee et al., 2015). Other HDAC inhibitors are currently under different phases of clinical trials for many cancers. These include abexinostat (Pharmacyclics), entinostat (Syndax Pharmaceuticals), and ricolinostat (Acetylon). HDAC inhibitors have been approved for several lymphomas and multiple myeloma. The lack of proper understanding of the mechanisms of action and specificities of the inhibitors hampers progress in this field. In addition, the lack of efficacy when used as single agents limits their potential as anticancer agents (Qiu et al., 2013). Combinations of HDAC inhibitors and immune checkpoint inhibitors are under trials for many cancers (Heninger et al., 2015).
Bromodomain and extra-terminal motif (BET) inhibitors
Bromodomain and extra-terminal motif (BET) proteins mediate many functions, including transcriptional regulation through binding to acetylated histone lysine residues (Pfister and Ashworth, 2017). Members of BET proteins include bromodomain-containing protein 2 (BRD2), BRD3, and BRD4 and their inhibition has been observed to stop the growth of many cancers, including breast, prostate, AML, and lymphoma (French, 2016). Inhibition of BET proteins has been shown to affect genes associated with super enhancers, multiple enhancers controlling cell type-specific genes (Hnisz et al., 2013; Pott and Lieb, 2015). Mitsubishi Tanabe Pharma created a BRD4 inhibitor named JQ1, which was found to displace the BRD4-NUT oncoprotein from chromatin and consequentially inhibits NUT midline carcinoma cell proliferation (Filippakopoulos et al., 2010).
BET inhibitors have been developed for different hematological malignancies and solid tumors and are under different phases of clinical trials. These include CP-0610 (Constellation Pharmaceuticals), FT-1101 (Forma Therapeutics), and BMS-986158 (Bristol-Myers Squibb) (Ali et al., 2017; Asangani et al., 2014; Markowski et al., 2017). Encouraging results from clinical trials are already filtering through with CP-0610 and others show effectiveness against cancer cells (Amorim et al., 2016; Stathis et al., 2016). Most BET inhibitors are under clinical trials as single agents, although others such as ZEN003694 and GS-5829 are used in combination with other therapies.
Histone methyltransferase (HMT) inhibitors
Histone methylation at different lysine residues can cause different effects on the expression of different genes, DNA replication, and repair (Wong et al., 2017). Histone H3 lysine 4 trimethylation (H3K4me3) is known to cause transcriptional activation, while histone H3 lysine 9 trimethylation (H3K9me3) leads to transcriptional repression (Li et al., 2017b; Zhang et al., 2016). It has been suggested that targeting individual histone methyltransferases (HMTs) may be more specific than DNMT and HDAC inhibitors (Becker et al., 2016).
Since the year 2000, when SUV39H1 gene was discovered, HMT inhibitors have been developed and are under trials (Castillo-Aguilera et al., 2017; Rea et al., 2000). Enhancer of Zeste homolog 2 (EZH2) promotes transcriptional silencing through methylation of histone H3 lysine 27 (H3K27) and it is known to be overexpressed in many cancers, including liver, lung, breast, and prostate cancers (Morin et al., 2010; Yap et al., 2011). EZH2 is known to promote carcinogenesis through increasing histone H3 lysine 27 trimethylation (H3K27me3) and through modulation of lineage-specific genes (Jin et al., 2017). EZH2 inhibitors, including GSK126, DS-3201b, tazemetostat (Epizyme), and CPI-1205 (Constellation), are under clinical trials (Knutson et al., 2013; Kondo, 2014).
EZH2 inhibitors work through blockage of H3K27 methylation and therefore inhibit activating mutations. GSK126 (GSK) is another EZH2 inhibitor under clinical trial for multiple myeloma and some solid tumors. All these EZH2 inhibitors are under trial as single agents.
DOT1L is an HMT for H3K79 and is important for the survival of leukemia cells that carry mixed lineage leukemia (MLL) gene translocations (Pfister and Ashworth, 2017). Inhibitors of DOT1L are known to selectively kill MLL-rearranged leukemia cells in mouse models (Daigle et al., 2013). Currently, several HMT inhibitors are under investigations, although some of them lack single-agent effects (Eggert et al., 2016).
HDM inhibitors
There are two subfamilies of HDMs and these are histone lysine demethylase 1 (KDM1) subfamily and the Jumonji C domain-containing subfamily. The catalytic domain of the Jumonji C domain-containing subfamily is highly conserved, making it very difficult to achieve a high degree of inhibitor selectivity (Hatch et al., 2017; Itoh et al., 2015). HDM inhibitors, including tranylcypromine, ORY-1001, and GSK2879552 (GSK), are undergoing clinical trials (Hatch et al., 2017). KDM1A is known to demethylate H3K4me and H3K4me2 and is overexpressed in breast, lung, and ovarian cancers (Hatch et al., 2017; Itoh et al., 2015).
Most importantly, the overexpression of KDM1A is associated with the downregulation of genes involved in immune response (Konovalov and Garcia-Bassets, 2013; Wei et al., 2018). Tranylcypromine induces apoptosis in leukemia cells, while GSK2879552 slows cellular growth in small cell lung cancer (SCLC) cells (Mohammad et al., 2015). Other members of the HDM family such as KDM4A and KDM4B are overexpressed in cancers, including esophageal, gastric, lung, and renal cancers (Berry and Janknecht, 2013). Inhibitors developed for KDM4A and KDM4B include NSC636819 and N-oxalylglycine (Chu et al., 2014; Hamada et al., 2009).
IDH inhibitors
IDHs are enzymes that catalyze the oxidative decarboxylation of isocitrate, and in the process produce α-ketoglutarate and carbon dioxide (Corpas et al., 1999; Yen et al., 2010). IDH mutations are found in cancers, including AML, gliomas, and thyroid cancers among others (Ragon and DiNardo, 2017). Specific inhibitors have been developed for IDH1 and IDH2 mutations and these include AG-120 (Agios), IDH305 (Novartis), and AG-221 (Agios). These inhibitors work through inhibition of mutant forms of IDH, and thus inhibit activating mutations. Both AG-120 and AG-221 are being studied as single agents and in combination with other epigenetic drugs such as azacitidine (Popovici-Muller et al., 2018).
Combination Therapy: Enhancing Efficacy of Cancer Drugs
Combination therapy is the gold standard when it comes to cancer treatment (Arseneau et al., 1974; DeVita, 2016). Few exceptions exist in terms of efficacy of monotherapy. Targeted tyrosine kinase inhibitors have shown high efficiency in treating patients with chronic myeloid leukemia (Druker et al., 2001). EGFR inhibitors also show increased tumor responses in cancers carrying certain mutations (Cancer Genome Atlas Network, 2015; Farshidfar et al., 2017; Huang et al., 2018). As with conventional drugs, regression of tumors can also be accompanied by the appearance of drug-resistant tumor cells (Baenke et al., 2016; Poulikakos and Rosen, 2011). In addition, similar to other therapies, individual patients respond differently to epigenetic drugs (Dzobo et al., 2018c). Thus, the future of epigenetic therapies relies on combining these therapies with conventional therapies as combination therapy.
Most epigenetic drug combinations are those of inhibitors of DNMT and HDAC involved in DNA methylation inhibition and histone deacetylation inhibition, respectively (Ahuja et al., 2016). Such combinations have been effective in reactivating epigenetically silenced genes and to cause apoptosis-mediated killing of cancer cells (Chai et al., 2008; Zhu et al., 2001). However, combining two or more drugs comes with the problem of potential high doses of drugs used than necessary. The consequential effect is increased off-target effects (Jones et al., 2016; Yuan et al., 2017).
Currently, many clinical trials are under way, which are utilizing combinations of inhibitors of DNMT and HDAC. So far, results have been conflicting with some clinical trials showing no beneficial effect of combining epigenetic drugs, while other studies have shown increased efficacy (Appleton et al., 2007; Garcia-Manero et al., 2017). Importantly, Phase I studies showed promising results with randomized Phase II trials showing no beneficial effect of combination therapy. Several randomized phase II trials showed no improved clinical outcome when DNMT and HDAC inhibitors were combined.
A combination of azacitidine and panobinostat in AML and MDS showed complete remission in about 27.5% of patients compared to 14.3% when drugs were used as single agents (Trial Number: NCT00946647) (Garcia-Manero et al., 2017). However, overall survival showed that there was no difference between monotherapy and combination therapy. In another combination trial, a combination of decitabine and valproic acid did not improve remission or overall survival of AML and MDS patients (Trial Number: NCT00414310) (Issa et al., 2015). A combination of azacitidine and vorinostat in patients with chronic myelomonocytic leukemia (CMML) showed overall response rate to be higher (38%) in monotherapy than in combination (27%) (Trial Number: NCT01522976) (Sekeres et al., 2017).
Suggested reasons for the lack of beneficial effect when combinations of epigenetic drugs are used include pharmacological antagonism. For example, azacitidine works well when there is cell division and DNA replication, while inhibitors of HDAC such as panobinostat and vorinostat promote cell cycle arrest and apoptosis. Inhibitors of HDACs are also not specific, resulting in broad effects on gene expression. Further research into the identification of potential biomarkers that can be used to identify patients who are likely to respond to individual and combination therapy is underway. Several clinical trials are registered at clinicalTrials.gov (https://clinicaltrials.gov) and show that epigenetic drugs are being trialled as single agents or in combination.
It has been shown that inhibitors of DNMT or HDAC when combined with other cancer therapies such as chemotherapy give rise to durable clinical response and increased efficacy (Benson et al., 2015). One possible explanation for this is that the epigenetic drugs sensitizes or primes cancers to chemotherapeutic drugs such as cisplatin (Borley and Brown, 2015; Fang et al., 2018). Indeed, several epigenetic mechanisms cause the development of resistance to chemotherapeutic drugs in many cancers (Sharma et al., 2010d). Currently, different combinations of epigenetic and chemotherapeutic drugs are under trials and results show increased efficacy (Fang et al., 2018; Matei et al., 2012).
Immune checkpoint therapy has been brought to the forefront of cancer therapy recently with the Nobel Prize in Medicine being awarded to immunotherapy pioneers James P. Allison and Tasuku Honjo. Many immunotherapy drugs have now been approved by the FDA and EMA for cancers such as renal, breast, and other cancers (Antonia et al., 2016; Planchard et al., 2016). Many cancers develop strategies to circumvent immune system detection and T cells are induced to tolerate cancer cells, hence there is no immune attack against cancer cells (Sharma and Allison, 2015; Topalian et al., 2015).
Recent data suggest that epigenetic mechanisms play a key role in tumor cell immune evasion (Dunn and Rao, 2017; Tomasi et al., 2006). The possible recruitment of HDACs to promoters of immune genes or increased methylation levels is likely to hinder antigen presentation, thereby influencing immune reaction (Khan et al., 2004; Magner et al., 2000). In addition, reduced recruitment of HAT CBP to gene promoters can result in faulty antigen processing mechanism (Setiadi et al., 2007). Indeed it has been observed that DNA methylation and other epigenetic mechanisms play huge roles in regulating T cell differentiation, the maturation of B cells, and the production of cytokines (Matheson and Corcoran, 2012; Tomasi et al., 2006).
Small-molecule inhibitors of enzymes responsible for epigenetic alterations are known to increase the ability of the immune system to respond to tumor cells (Coulie et al., 1999; Fonsatti et al., 2007). Epigenetic drugs and immunotherapy have been combined for patients with nonsmall cell lung cancer with results showing durable responses (Ikeda et al., 2016; Wrangle et al., 2013). Many studies are underway to understand how epigenetic drugs help immunotherapy (Doi et al., 2017; Wu et al., 2018).
One suggested reason why epigenetic drugs work well with immunotherapy is that epigenetic drugs are thought to reverse immune evasion by inducing the expression of surface proteins needed for identification of cancer cells by immune cells (McCaw et al., 2018). Furthermore, the efficacy of many immunotherapeutic compounds has been increased by the presence of small-molecule inhibitors of enzymes responsible for epigenetic modifications (Karpf, 2006; Srivastava et al., 2014). Decitabine and 5-azacytidine are known to induce major histocompatibility complex (MHC) gene expression and receptors on natural killer cells (Kopp et al., 2013; Serrano et al., 2001).
The ability of T cells to kill breast, lung, and colon cancer cells was increased by the presence of HDAC inhibitors (Gameiro et al., 2016). EZH2 inhibitors induced increased CXCL9 and CXCL10 chemokine production resulting in the recruitment of T cells. T cells were able to kill cancer cells in a xenograft model (Nagarsheth et al., 2016). Azacitidine has been shown to induce interferon signaling in lung cancer cells and this results in increased PD-L1, sensitizing cancer cells to immune checkpoint blockade in the process (Kelly and Issa, 2017).
However, another study showed that the use of EZH2 inhibitors caused a decrease in CD4+ T cells with the possibility of preventing T cell-mediated cancer cell destruction (Tong et al., 2014). JQ1, an inhibitor of BET, is known to reduce PDL1 expression and slow tumor growth through activating T cells (Zhu et al., 2016). On the other hand, DNMT inhibitors are known to increase PDL1 expression in leukemic cell lines (Wrangle et al., 2013; Yang et al., 2014). Anti-PD1, for example, can be combined with DNMT inhibitor such as azacitidine to increase tumor regression in a xenograft model (Kim et al., 2014). Furthermore, azacitidine in combination with T cells derived from patients resulted in more killing of tumor cells (Kunert et al., 2016). GSK126, an EZH2 inhibitor, can also increase the efficacy of immune checkpoint inhibitors (Peng et al., 2015).
These data support the notion that epigenetic inhibitors can act in concert with conventional therapies and immunotherapy to increase the efficacy of cancer treatment. Clinical trials are underway to evaluate the utility of combining inhibitors of HDACs and DNMTs with immune checkpoint inhibition (Adra et al., 2018). Pembrolizumab, an immune checkpoint inhibitor, together with azacitidine showed higher tumor regression in relapsed Hodgkin lymphoma (Falchi et al., 2016; Pfister and Ashworth, 2017). Pembrolizumab, nivolumab, ipilimumab, and durvalumab are the immunotherapy drugs most used in combination with epigenetic drugs for several malignancies. To fully elucidate how epigenetic drugs augment immunotherapy, it is important to understand how these drugs affect immune cells as well.
Combination of different epigenetic therapies, including small molecules combined with epigenetic drugs, is under trial. Many pharmaceutical companies, including Agios, GlaxoSmithKline plc, and Novartis, are spearheading these combinatorial studies with results expected to help design future studies. Results so far show that different epigenetic drug combinations can induce antitumor responses (Zou et al., 2017).
A combination of epigenetic drugs can also be used to sensitize cancer cells before the use of immune checkpoint therapy. Inhibitors of both DNMT and EZH2 are known to cause the overexpression of CXC-motif chemokine 9 (CXCL9). CXCL9 together with CXCL10 stimulate T helper 1 cells (Jones et al., 2016; Peng et al., 2015). Combinations of inhibitors of BET and HDAC and other therapies are already under clinical trials and many such studies are likely to position epigenetic therapy at the forefront of cancer treatment.
One area that has been receiving attention lately is the role of epigenetics in cancer therapy resistance. It is known that some cancer cells might adopt an epigenetic-mediated chemoresistance, showing a repressed chromatin state (Sharma et al., 2010d). Trichostatin A, an HDAC inhibitor, and other epigenetic inhibitors are known to inhibit these cancer cells with an epigenetic-mediated chemoresistance (Harmeyer et al., 2017). Unfortunately, cancer cells can also develop resistance to epigenetic inhibitors (Rathert et al., 2015). Combining epigenetic inhibitors and conventional drugs may offer a durable drug response (Fig. 5).
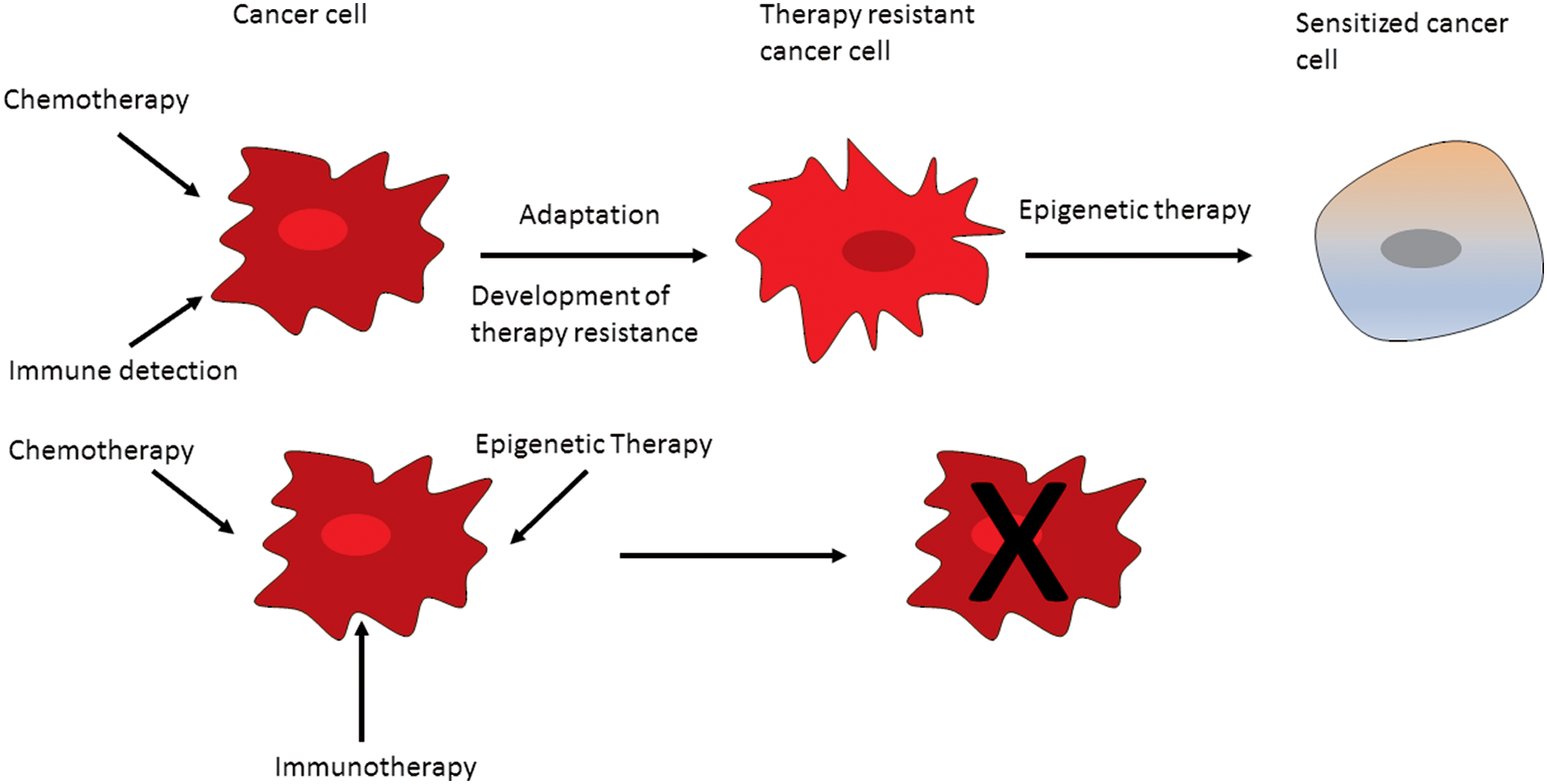
Therapy can provide a selection pressure leading to both genetic and epigenetic evolution. Ultimately, there is the emergence of resistant tumor cells.
Future Perspectives
To exploit the potential of epigenetic drugs, it is important that novel applications of epigenetic inhibitors be found such as in immune-oncology. Drug screening assays have made use of cell lines coupled with knockdowns of specific proteins using mostly siRNA or shRNA (Chan and Giaccia, 2011). Coupled to the development of better disease models such as 3D organoids is the need for new epigenetic editing tools (Clevers, 2016). The editing of the epigenome components can unravel their contribution to disease initiation and development, and can be used to correct certain mutations driving certain diseases. Pathological conditions resulting from the overexpression or low expression of a gene/genes may benefit from epigenetic editing, allowing the downregulation or upregulation of such a gene/genes, ultimately correcting the phenotype of a cell.
The advent of novel gene editing tools such as clustered regularly interspersed short palindromic repeat (CRISPR)-Cas9 and transcription activator-like effector nuclease (TALEN) has revolutionized the field with few off-target effects and increased reproducibility of results (Chiang et al., 2016; Manguso et al., 2017). Even better is the ability of CRISPR-Cas9 technology to silence and activate specific genes (Du and Qi, 2016). Looking ahead, gene editing technologies such as CRISPR-Cas9 and TALEN are likely to greatly influence the discovery of new drug targets that might be translated into the clinic. Novel technologies such as CRISPR-Cas9 and APOBEC-coupled epigenetic sequencing (ACE-seq) are likely to dramatically spur epigenetic research and ultimately its clinical applications.
As described by Schutsky et al. (2018), ACE-seq is a method that does not utilize bisulfite and localizes 5-hydroxymethylcytosine at single-base resolution. This is achieved with very low concentration of DNA input. The gold standard epigenetic assay used for the detection of methylated cytosine is the bisulfite treatment of DNA followed by PCR amplification (Adusumalli et al., 2015; Lee and Smallwood, 2018). In standard bisulfite sequencing, cytosine is converted to uracil, but leaves 5mC and 5hmC unconverted (Lee and Smallwood, 2018; Yu et al., 2018). Other sequencing methods such as oxBS-seq and TAB-seq are coupled to bisulfite to differentiate 5mC and 5hmC bases (Yu et al., 2018).
However, this bisulfite treatment requires the use of extreme pH and elevated temperatures. This chemical treatment can destroy the DNA sample. The new method, ACE-seq, is an enzymatic method utilizing one member of the AID/APOBEC family of enzymes that catalyzes the deamination of cytosine to uracil in single-stranded DNA (Schutsky et al., 2018). One of the most appealing factors of the new method, ACE-seq, is that it can be used to sequence the whole genome.
In 2017, Liao et al. (2017) described a CRISPR-Cas9-based method for activating target genes in vivo through transepigenetic remodeling. At the moment, genome editing methods require the induction of DNA double-strand breaks that can cause mutations, limiting the utility of current gene-editing methods in clinical therapies (Liao et al., 2017; Su et al., 2016). In addition, the use of CRISPR-Cas9 system in editing the epigenome has not succeeded in producing relevant phenotypes for research (Jurkowski et al., 2015; Saunderson et al., 2017). The novel method can be used to modulate histone modifications and not DNA sequences. This system was able to ameliorate phenotypes associated with human diseases in mouse models.
Before the advent of this method, most strategies used to alter epigenetic processes utilized small-molecule inhibitors that would add or remove histone modifications. This method has the potential to affect off-target genes. These two technologies, ACE-seq and CRISPR-Cas9, will allow scientists to answer many unresolved questions about the role of epigenetics in causing changes in gene expression. The two methods can also help investigate the potential synergistic activities of different epigenetic modifications and also how epigenetic modifications affect other mechanisms of gene control such as transcription factors.
Furthermore, these novel technologies can also allow scientists to answer questions such as how environmental factors do affect gene expression through epigenetic mechanisms to produce cells with a certain phenotype. Most importantly, using disease models, scientists will be able to determine how small-molecule inhibitors of epigenetic modifications or other drugs can reverse epigenetic changes. By answering these important questions, epigenetics will become incorporated into disease diagnosis and treatment strategies.
Companies, including Novartis, AstraZeneca, GSK, and Gotham Therapeutics and Epigenomics have launched many initiatives into epigenetic research with the hope of finding drugs that can cure many pathological conditions such as cancer, Alzheimer's disease, and other diseases (Kurtz et al., 2017; Ribich et al., 2017). Research into the role of epigenetics in human health and disease has been expanded into other human conditions such as aggression, happiness, depression, and risk taking.
Indeed, epigenetics has entered a new exciting era and scientists and clinicians cannot wait for answers to some of unanswered human conditions. In addition, the model used in the genetic and drug screens is likely to improve over time. Lately, 3D organoids have changed the landscape of tissue modeling, having advantages over conventional models such as cell lines and xenografts (Clevers, 2016; Dutta et al., 2017; Dzobo et al., 2018b).
Genome-wide data deposited within public and private domains contain research on the use of siRNA, shRNA, and CRISPR-Cas9 screens done on many cell lines, and lately on organoids as well (Deans et al., 2016; Hart et al., 2017). Websites contain data from studies on DNA methylation and chromatin, for example. In addition, the Sanger Institute has data from a screen of many cancer cell lines and anticancer compounds (Giovannetti et al., 2012; Ye et al., 2018). There are a lot of tools and databases that are essential and useful for scientists working on the epigenome.
For example, the International Human Epigenome Consortium database contains reference epigenomes for different diseases and other conditions (https://epigenie.com/epigenetic-tools-and-databases). Other consortiums and reference epigenomes include Roadmaps Epigenomics, CEEHRC Platform, DeepBlue, and The Epigenome Atlas. Epigenetic databases include 4DGenome, Histome, and cisRED. There are several data analysis and visualization packages available for scientists. Beside the use of the statistical program R, other packages include Bsseq, epiGbs, MethPipe, and DMAP.
Conclusions
In order for cancer cells to maintain their uncontrollable growth and evade detection, both the genome and epigenome are aberrantly changed. Light microscope evidence during pathological diagnosis has shown over the years that the chromatin of cancer cells is different from that of normal cells. The role of epigenetic alterations in cancer cell ability to prevent detection and develop therapy resistance is likely to be bigger than is known. Both mutations in important genes and the plasticity of epigenetic changes are likely to provide cancer cells with a “fertile” phenotype to evolve and become therapy resistant.
While drug discovery has mostly focused on factors such as growth factors and signaling proteins, the epigenome is receiving much attention and is likely to change the face of drug discovery forever. The likelihood of epigenetically reprogramming cancer cells to normal cells or at least prime cancer cells to other therapies appears certain. In this regard, the use of next-generation sequencing and gene editing technologies will likely increase our understanding of epigenetic control in cancer leading to increased transformation in cancer treatment. In agreement with the idea that cancer treatment should be at best combinatorial in nature, epigenetic drugs are likely to be effective when used in combination with other therapies such as chemotherapy and immunotherapy.
Footnotes
Acknowledgments
Funding was provided by the International Centre for Genetic Engineering and Biotechnology (ICGEB), the National Research Foundation (NRF) of South Africa, the Next Einstein Fellow Program, and the University of Cape Town (UCT). The funders had no role in the conduct of the research or the preparation of the article. I would like to thank Prof Vural Özdemir for editorial comments.
Author Disclosure Statement
The author declares that there are no conflicting financial interests.