
Abstract
Abstract
Historically, plant biology studies have lagged behind systems biology studies in animals and humans. However, there are signs of positive change as evidenced by the rise of big data in plant proteomics, and the availability of data science tools and next-generation sequencing technologies. Currently, the sequence information on nearly 300 plant species is available although they are curated to varying degrees of sophistication. This has led to significant enrichment of representations in the corresponding plant proteome databases. Analysis of the proteome component of an organism offers structural, functional, and network scale insights. Moreover, the development of high-throughput mass spectrometric techniques has augmented our understanding of proteins and their expression patterns under various conditions. Several thousand proteins can now be identified from a single mass spectrometric analysis. In this expert review, we provide an in-depth analysis on plant proteome databases, how to access them, and, importantly, the biological, research, and application contexts in which each database is significant, their comparative strengths, and limitations. We aimed in this analysis to reach out to young scholars embarking on plant biology and proteomic research as well as to those already established in the field so as to provide integrated critical analyses of plant proteome databases and bioinformatics tools in this nascent field of systems sciences. In conclusion, plant proteome research is an emerging and exciting frontier of integrative biology scholarship and innovation. Our future efforts must also be invested in integrating the available databases to allow for multiomics data analysis, research, and development.
Introduction
The advent of next-generation sequencing platforms has led to generation of massive amounts of biological big data. In addition, high-throughput mass spectrometric techniques have further amplified the amount of available proteomic data. The article “A draft map of the human proteome” has recently reported the identification of proteins encoded by 17,294 genes (Kim et al., 2014).
Plant proteome research is an exciting and essential frontier of integrative biology. The present expert review offers a deeper understanding of the plant proteome databases, how to access them, and, importantly, the biological, research, and application contexts in which each database is significant, their comparative strengths, and limitations (Fig. 1).
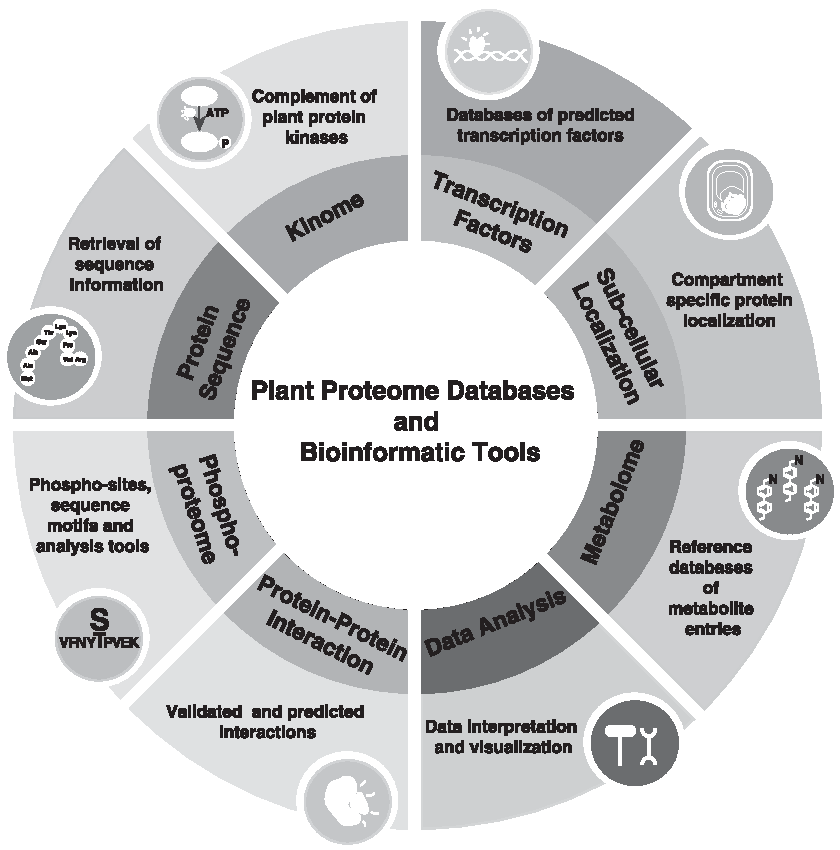
Schematic representation of the plant proteome databases and bioinformatics tools useful for plant proteome analysis.
We aimed to reach out to young scholars embarking on new careers in plant systems biology and proteomic as well as those already established in the field so as to provide integrated critical analyses of plant proteome databases and bioinformatics tools in this nascent field of systems sciences.
Dawn of Proteomics Big Data
Proteomics big data are now emerging in plant research as well. Proteome analysis in plant systems has been carried using various tissue samples such as leaves, root, flower, and fruits among others (Feng et al., 2017; Hopff et al., 2013; Jia et al., 2017; Szymanski et al., 2017). Initially, many studies have been carried out using two-dimensional electrophoresis (2-DE) gel-based techniques. For example, the SWISS-2DPAGE (http://world-2dpage.expasy.org/swiss-2dpage/) houses data related to 2-DE gels, including that of Arabidopsis thaliana, wherein one can locate a protein from UniProtKB/Swiss-Prot on the gel. However, the last update for the database was made in 2011.
Advancements made in chromatographic techniques together with improved resolution powers of mass spectrometers are now paving the way for gel-free techniques of plant proteome analysis (Takac et al., 2017; Tan et al., 2017). Subcellular proteomics have been carried out to gain comprehensive understanding of the compartment-specific data (Albenne et al., 2013; Lee et al., 2013; Narula et al., 2013). Such data sets can be queried for experimental evidence about protein subcellular location (Hooper et al., 2017).
In addition to proteome analysis, high-resolution mass spectrometers allow the analysis of protein post-translational modifications. In this context, data have been recently accumulating, for example, on phosphorylation, acetylation, and succinylation (Hartl et al., 2017; Kumar et al., 2017; Zhen et al., 2016). Information about sites of modifications is documented in databases such as The Arabidopsis Protein Phosphorylation Site Database (PhosPhAt), PHOsphorylation SIte DAtabase (PHOSIDA), and Plant Protein Phosphorylation DataBase (P3DB) for phosphorylation, eukaryotic Writers, Erasers and Readers protein of Histone Acetylation, and Methylation system Database (WERAM) for histone acetylation and methylation (Durek et al., 2010; Gao et al., 2009; Gnad et al., 2011; Zulawski et al., 2013).
Apart from these dedicated databases, dbPTM provides access to available data on various post-translational modifications and OMICtools allows users to navigate various resources and prediction tools for analysis of post-translational modifications (Henry et al., 2014; Huang et al., 2016). High-throughput proteomic studies generate large volumes of data in the order of 15–20 GB per day.
The ProteomeXchange consortium was created to facilitate storage and dissemination of mass spectrometry-derived data. Data can be submitted to the public data repository Proteomics IDEntifications (PRIDE) database (https://www.ebi.ac.uk/pride/archive/). It serves as a “go to” resource for researchers to archive available data for (re)analysis.
The selection of starting materials for proteomics studies is the most crucial step in conducting an experiment. Databases such as the phenotype database of Arabidopsis mutant traits AraPheno and RARGE enable selection of plants based upon the reported phenotypes (Akiyama et al., 2014; Lloyd and Meinke, 2012; Seren et al., 2017).
The Human Protein Atlas and the Human Protein Reference Database (HPRD) house human protein information including, but not limited to, their tissue and subcellular localization, expression and association with diseases, post-translational modification, and protein–protein interactions (Keshava Prasad et al., 2009; Uhlen et al., 2015; Thul et al., 2017). The wealth of information in Arabidopsis is available in The Arabidopsis Information Resource (TAIR).
Protein Sequence Databases
Sequence information for most of the sequenced plant genomes can be retrieved from databases such as NCBI Viridiplantae, GenBank, DDBJ, and UniProt that host related protein sequence information in addition to nucleotide sequence information. Sequence retrieval can also be performed from databases dedicated to plants such as Phytozome, plaBi, and Gramene database, which are subsets of the EnsemblPlants database and PlantGDB.
Dedicated databases for individual plant species are available such as TAIR for A. thaliana, TOMATOMICS for Solanum lycopersicum, maize GDB for Zea mays, Rice Genome Annotation Project for Oryza sativa ssp. japonica cv. Nipponbare, legume-specific protein database LegProt, and the wheat proteome database (Duncan et al., 2017; Harper et al., 2016; Kawahara et al., 2013; Kudo et al., 2017; Lei et al., 2011; Reiser et al., 2017). The list of databases available for plant proteome analysis is given in Table 1.
Databases Essential for Plant Proteome Analysis
BLAST, basic local alignment search tool; GO, gene ontology; MRM, multiple reaction monitoring; MS/MS, mass spectrometry/mass spectrometry (tandem mass spectrometry); PTMs, post-translational modifications; TAIR, The Arabidopsis Information Resource.
Plant Kinase Databases
The model plant Arabidopsis has twice as many protein kinases as compared with the human complement (Zulawski et al., 2014). With a total of 1052 kinases and 162 phosphatases (Wang et al., 2014a), the numbers are still counting, making it important to characterize the roles of these signaling components in a context-dependent manner.
After the first publication related to the classification of Arabidopsis kinases (Shiu and Bleecker, 2001), more kinases were subsequently added to the list (Wang et al., 2014a; Zulawski et al., 2014). Sequence information of Arabidopsis kinases can be retrieved from sources such as the database PlantsP (Tchieu et al., 2003). The database contains information related to 979 unique Arabidopsis kinases and 125 phosphatases classified into various groups. Few other kinases from other plant species are also included. The database has, however, not been populated with the newly identified kinases. The complement of Arabidopsis kinases is also available in P3DB (1186 entries) although not downloadable as a single list.
Perhaps the most comprehensive list of Arabidopsis kinases is currently available at EKPD the Eukaryotic protein Kinases and protein Phosphatases Database (Wang et al., 2014a). This database was updated in 2013 and currently contains protein kinases and protein phosphatases from 84 eukaryotic species. They are further divided into groups and classes based upon their catalytic domains. The information is accessible to the public and it can be downloaded.
Information from 22 plant species (A. thaliana, Arabidopsis lyrata, Brassica rapa, Glycine max, Populus trichocarpa, S. lycopersicum, Solanum tuberosum, Vitis vinifera, Brachypodium distachyon, Hordeum vulgare, Musa acuminate, Oryza glaberrima, Oryza indica, O. sativa, Oryza brachyantha, Setaria italic, Sorghum bicolor, Z. mays, Selaginella moellendorffii, Physcomitrella patens, Chlamydomonas reinhardtii, and Cyanidioschyzon merolae) has been included. For Arabidopsis, 1052 kinases and 162 phosphatases are categorized into groups. The protein and/or domain sequences can be downloaded from the advanced search option, wherein the sequences are arranged according to the organisms. The databases containing information related to plant kinases are listed in Table 1.
Plant Transcription Factor Databases
Some of the available plant transcription factor databases have earlier been reviewed and compared (Mitsuda and Ohme-Takagi, 2009). The database of Arabidopsis transcription factors (DATF) (Guo et al., 2005) is linked to ATRM: Arabidopsis Transcriptional Regulatory Map, which is a subset of the PlantTFDB (Jin et al., 2014). The latter contains information about transcription factor of >80 plant species.
The Arabidopsis transcription factor database (AtTFDB) houses a large collection of Arabidopsis transcription factors that can be fetched using either the locus ID or gene names (Yilmaz et al., 2011). The Arabidopsis transcription factors can also be browsed according to their gene families. As the database was created using TAIR9, revisions in the TAIR10 database (if any) need to be incorporated. Another database of Arabidopsis transcription factors is the RIKEN Arabidopsis Transcription Factor database (RARTF) (Iida et al., 2005). A comprehensive collection of biotic and abiotic stress responsive putative transcription factors from Arabidopsis and rice (O. sativa subsp japonica and O. sativa subsp indica) is available at Stress Responsive Transcription Factor Database (STIFDB V2.0) (Shameer et al., 2009).
The Arabidopsis transcription factors are linked to the source references, however. They have not been updated since 2012. The LegumeTFDB houses information of G. max, Lotus japonicas, and Medicago truncatula Arabidopsis transcription factors (Mochida et al., 2010). The Cicer Transcription Factor Database (CicerTransDB) has recently been developed for the legume crop Cicer arietinum L. (Gayali et al., 2016). Other legume-specific TF databases include pigeonpea PpTFDB and Phaseolus vulgaris PvTFDB (Bhawna et al., 2016; Singh et al., 2017).
Database resources for other plants include wDBTF for wheat, FmTFDb for foxtail millet, and TreeTFDB a database of the transcription factors from six economically important trees (Bonthala et al., 2014; Mochida et al., 2013; Romeuf et al., 2010). Although information related to plant Arabidopsis transcription factors is available, variations in the absolute numbers and classification systems are an evidence for the need toward more sophisticated curation. The databases containing information related to plant transcription factors are listed in Table 1.
Plant Organellar Proteomics Databases
The SUBA (SUBcellular location of proteins in Arabidopsis) database serves to coalesce information on protein localization from large-scale organellar proteomics and green fluorescence protein (GFP) localization studies conducted in Arabidopsis (Hooper et al., 2017). The database includes data sets based on various protein localization prediction tools and data retrieved from Swiss-Prot annotation.
The current version SUBA4, the bibliographic references having been last updated in June 2016, houses information for nearly 60,000 experimental protein location claims. The feature SUBAcon determines the consensus location of query proteins from experimental and in silico predictions.
The Plant Proteome Database (PPDB) houses data from organellar proteome studies (Sun et al., 2009). Users can access lists of proteins by choosing a function, biochemical pathway, subcellular location, or a post-translational modification. Eukaryotic Subcellular Localization DataBase (eSLDB) contains the protein subcellular localization information of eukaryotic organisms (Pierleoni et al., 2007). A nonredundant list of 30,600 Arabidopsis proteins is available along with their localization information, if any. However, most of the data available are based upon in silico analysis and the data have not been updated.
ARAMEMNON catalogs the Arabidopsis membrane proteins. The current release includes proteins from nine plant species (A. thaliana, V. vinifera, P. trichocarpa, S. lycopersicum, Cucumis melo, O. sativa, Z. mays, B. distachyon, and M. acuminate) (Schwacke and Flugge, 2018). Features associated with membrane proteins such as probable lipid modifications [glycosylphosphatidylinositol (GPI)-attachment, prenylation, myristoylation, and palmitoylation], details of transmembrane spanning regions (alpha helices and beta barrel positions) and their subcellular location can be displayed based on the outputs of various prediction tools. A dedicated section is reserved for membrane transporters, referred to as plant “permeome.” Although well curated, the database does not include data sets from large-scale mass spectrometric analysis that would aid in enriching the number of representations.
The Arabidopsis Nucleolar Protein database (AtNoPDB) consists of nucleolar proteins identified through proteomics analysis (Brown et al., 2005). The data set has been compared with that of the human complement (Andersen et al., 2005). Although well structured, the incorporation of currently available data from Arabidopsis and other plant species would make it more useful (Gonzalez-Camacho and Medina, 2004; Palm et al., 2016).
The AT_Chloro database was constructed for indexing the Arabidopsis chloroplast proteins identified through mass spectrometry. The knowledge base is enriched by the incorporation of data from three highly enriched subplastidial regions envelope, stroma and thylakoids and subthylakoidal regions grana and stroma-lamellae (Bruley et al., 2012; Ferro et al., 2010; Tomizioli et al., 2014). The AT_CHLORO database is the first accurate mass and time database dedicated to plants. The database is also linked to other databases that allow users to confirm the annotation of query protein(s).
The database was last updated in the year 2015. The mitochondrial proteome of Arabidopsis is available in two different sites, both referred to as the Arabidopsis Mitochondrial Protein Database. In the former, orthologous proteins are compared across a diverse range of organisms to demonstrate the divergence of the plant mitochondrial proteome. The latter contains information gathered from small-scale proteome studies such as 2-DE analysis and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The WallProtDB has been created as a resource for plant cell wall proteomics (San Clemente and Jamet, 2015). The list of databases containing information related to protein subcellular localization is given in Table 3.
Databases, Tools, and Resources for Functional Proteomics Analysis
3D, three-dimensional.
Plant Phosphoproteomics Databases
The Arabidopsis Protein Phosphorylation Site Database (PhosPhAt 4.0) currently houses one of the largest collections of protein phosphorylation sites identified through mass spectrometry (Durek et al., 2010). The database is presented as a web application to enable data retrieval. The availability of annotated spectra of phosphopeptides makes it useful for designing targeted proteomics analysis and data verification. The database allows users to access curated information from kinase–target interactions for several kinases and phosphatases (Zulawski et al., 2013). Data from mass spectrometric studies as well as from kinase–substrate interaction studies can also be submitted to the database.
The P3DB initiated with protein phosphorylation data of oilseed rape was later expanded to integrate data from other plant species (Gao et al., 2009). Currently, P3DB 3.0 hosts phosphosites belonging to 16, 477 phosphoproteins. A list of 1186 plant kinases and 159 phosphatases is available in the database. A Basic local alignment search tool (BLAST) utility option is also available to retrieve orthologous phosphoproteins or phosphopeptide sequences. A link to the Musite website is provided to predict the site of phosphorylation in the query sequence. Other extended data include the protein domain, protein–protein interaction, and ontology information along with kinase (or phosphatase)–substrate information Kinase Client Assay (KiC Assay) data, if available (Yao and Xu, 2017; Yao et al., 2014). Users can also choose to submit their data into the respective experimental categories.
The database of Phospho sites in PlanTs (dbPPT) serves as an integrated resource for protein phosphorylation events reported in plants. It contains both manually curated and information from other public databases, including PhosPhAt (Durek et al., 2010) and P3DB (Gao et al., 2009). The database was launched in 2014 (Cheng et al., 2014) and information related to several thousand phosphorylation sites belonging to 20 plant species is currently available (dbPPT 1.0), making it the most comprehensive collection of plant phosphorylation sites to date.
Available data can be retrieved using various search options such as the organism, function, gene name, protein name, taxa ID, UniProt accession number, reference source database, and sequence source database. The phosphopeptide sequence along with the site of phosphorylation, the database source, and the associated reference is provided. Protein sequence along with the sequence annotations and domain information is embedded in the output. The search speed is, however, rather slow. It would be more useful if the data were available in a tabulated downloadable format. The MedicagoPhosphoProtein Database contains mass spectrometry-derived phosphoproteome data of M. Truncatula, a major model legume (Grimsrud et al., 2010). The databases containing information on plant phosphoproteome are listed in Table 2.
Databases and Tools Essential for Post-translational Modification Analysis
FASTA, FAST-All.
Protein Structure and Domain Databases
Few websites such as CYBIONIX and Bioinformatics Software and Tools are particularly useful as they contain useful collections of links to retrieve sequence information as well as to various analysis tools. The Protein Data Bank is an integrated resource of protein structures and other related information. A list of databases and tools for proteomics data analysis is given in Table 3.
Protein–Protein Interaction Databases
The in vivo interactions between protein scan define the set of plant-specific functions. A set of 6200 binary interactions among ∼2700 proteins has been catalogued by the Arabidopsis Interactome Mapping Consortium (Consortium, 2011). Such interactions are also documented in the IntAct Molecular Interaction Database (Orchard et al., 2014). Recently PPIM, a protein–protein interaction database, was created for maize (Zhu et al., 2016). The database Computational System Biology houses a list of plant protein–protein interaction databases such as PPIM and the Database of Interacting Proteins in Oryza sativa (DIPOS) (dedicated to protein interactions in rice).
A. thaliana Protein Interactome Database (AtPID) is a resource wherein protein interactions are integrated with genotype–phenotype associations of Arabidospsis mutants (Cui et al., 2008). The Plant Interactome project hosted by the Salk Institute intends to document protein–protein interactions of Arabidopsis using yeast-2-hybrid and protein array technologies (http://signal.salk.edu/interactome.html).
The STRING database can be used to query protein interactions from multiple species including Arabidopsis. The data include both predicted and those with experimental evidence. Biological General Repository for Interaction Datasets (BioGRID) contains a large collection of protein–protein interactions from several organisms (Chatr-Aryamontri et al., 2017).
The database can be queried using the gene identifier and displayed results include the protein's gene ontology (GO) category and publication from where the information is retrieved. AIM, Arabidopsis Interactome Modules Database, aims at identifying the protein constituents within interactome modules (Wang et al., 2014b). The data are linked to gene expression data sets derived from microarray analysis. The database is also intended to catalog interlogs in various other plant species. GeneMANIA contains data sets from other interactome databases and it enables the user to predict the functions of genes based upon their protein–protein interactions (Warde-Farley et al., 2010). Databases and tools related to plant protein–protein interactions are listed in Table 3.
Computational Tools for Predicting Post-Translational Modifications (PTMs)
Computational tools such as DISPHOS, NetPhosK, KinasePhos, and scan-X have been widely used for predicting protein phosphorylation sites. DISPHOS (www.dabi.temple.edu/disphos/) prediction is based upon the disorder region surrounding the phosphorylation site, whereas NetPhosK allows the predictions of kinase-specific eukaryotic protein phosphorylation sites (Blom et al., 2004). KinasePhos utilizes sequence-based amino acid coupling-pattern analysis and solvent accessibility (Huang et al., 2005). The scan-X tool can predict the kinase recognition motifs within a phosphoproteome data set (Chou and Schwartz, 2011). The tools are, however, generic in nature and have mostly been trained using from human phosphoproteome data sets.
Few tools have been designed for the analysis of phosphorylation sites on plant proteins. Musite is an application that allows the prediction of protein phosphorylation sites using local sequence similarities, protein disorder scores, and amino acid frequencies (Gao et al., 2010). The current version (Musite 1.0) can perform the analysis for 6 eukaryotic organisms including A. thaliana and kinase-specific prediction models for 13 kinases or kinase families. The application can also be downloaded as an open-source standalone tool. The PhosPhat database also has a built-in plant-specific phosphorylation site prediction tool trained to predict phosphorylation on Ser, Thr, and Tyr residues (pSer, pThr, and pTyr).
PlantPhos is a web tool developed for the prediction of phosphorylation sites on plant proteins based upon the recognition of kinase motifs on the substrates (Lee et al., 2011). This tool has been trained using the experimental Arabidopsis phosphorylation data available on TAIR9 database. Significantly conserved motifs are clustered based upon the maximal dependence decomposition. The Rice_Phospho 1.0 was developed to predict protein phophorylation sites in rice (Lin et al., 2015). Bioinformatic tools for predicting other post-translational modifications (PTMs) include computational tools for prediction of lysine acetylation (Basu, 2013; Deng et al., 2016) and prediction of ubiquitination sites (Chen et al., 2015; Walton et al., 2016). The list of software tools for performing PTM prediction on plant proteins is given in Table 2.
Other Computational Tools
The OMIC tools contain a suite of links to various software tools that are useful for proteomics data interpretation (Henry et al., 2014). The newly developed platform ePlant allows users to seamlessly navigate to available data on Arabidopsis, including genome, proteome, interactome, transcriptome, and three-dimensional molecular structure data (Waese et al., 2017). Along with download option, it is also useful data for data visualization. The Arabidopsis Information Portal (Araport) is a resource developed for plant biology with special emphasis on integrative data analysis (Krishnakumar et al., 2015). ThaleMine developed by the Araport team provides access to data available on Arabidopsis such as RNA-seq and array expression, coexpression, protein interactions, homologs, pathways, publications, alleles, germplasm, and phenotypes (Krishnakumar et al., 2017).
Another tool that allows integrative data analysis is the CORNET (CORrelation NETworks). The platform allows access to coexpression data, protein–protein interactions, regulatory interactions, and functional annotations (Van Bel and Coppens, 2017). The Arabidopsis Proteotypic Predictor is a web-based tool that enables users to select candidate transitions for Selected Reaction Monitoring (Taylor et al., 2014). Another such web-based tool is MRMaid that serves as resource for transitions for a subset of Arabidopsis proteins (Fan et al., 2012). Tools for molecular network visualization and data integration include Cytoscape and all the plugins contained therein (Killcoyne et al., 2009).
Future Perspectives
The dawn of big data in proteomics is changing the landscape of systems biology research practices. Both emerging and established investigators need access to a wide a range of databases in plant proteomics, together with the knowledge of their comparative strengths and limitations. The present review has attempted to address this knowledge gap in the field.
It is noteworthy that plant biology has historically and markedly lagged behind systems biology studies in animals and humans. However, there are signs of positive change as evidenced in the present review and the rise of big data in plant proteomics. Still, the data submitted to public data repositories for public access remain scattered across various databases and websites. There is a need for well-designed and curated protein databases akin to the HPRD and the Human Protein Atlas.
Also, there exists a severe shortage of plant databases for biological pathways such as NetPath (Kandasamy et al., 2010). Detailed knowledge of the stress signaling pathways and those involved in plant secondary metabolite synthesis is important to target molecular components for improved stress tolerance or yields. Finally, we suggest that our future efforts must also be invested in integrating the available databases to allow for multiomics data analysis, research, and development in the field of plant biology.
Footnotes
Acknowledgments
The authors thank Yenepoya (Deemed to be University) for full access to the instrumentation facility. P. S. is funded by the Early Career Research Award (Award no.: ECR/2016/000365) from Science & Engineering Research Board SERB), Government of India. C.N.K. is a recipient of Junior Research Fellowship in the OLAV THON Foundation funded grant at Yenepoya.
Author Disclosure Statement
The authors declare they have no financial conflicts of interest.