
Abstract
Abstract
Pharmaceutical industry and the art and science of drug development are sorely in need of novel transformative technologies in the current age of digital health and artificial intelligence (AI). Often described as game-changing technologies, AI and machine learning algorithms have slowly but surely begun to revolutionize pharmaceutical industry and drug development over the past 5 years. In this expert review, we describe the most frequently used machine learning algorithms in drug development pipelines and the -omics databases well poised to support machine learning and drug discovery. Subsequently, we analyze the emerging new computational approaches to drug discovery and the in silico pipelines for drug repositioning and the synergies among -omics system sciences, AI and machine learning. As with system sciences, AI and machine learning embody a system scale and Big Data driven vision for drug discovery and development. We conclude with a future outlook on the ways in which machine learning approaches can be implemented to buttress and expedite drug discovery and precision medicine. As AI and machine learning are rapidly entering pharmaceutical industry and the art and science of drug development, we need to critically examine the attendant prospects and challenges to benefit patients and public health.
Introduction
Pharmaceutical industry specifically and the art and science of drug development broadly are sorely in need of novel transformative technologies in the current age of digital health and artificial intelligence (AI) (Kohane, 2015).
A brief description of the history of AI has been recently discussed by Garvey (2018) in terms of three paradigms: “GOFAI” (1950-60s), “Expert Systems” (late 1970-80s), and “machine learning” (2010-present). The first, GOFAI, short for “good-old-fashioned AI”, has focused on the creation of general purpose logic systems and led to the development of foundational techniques such as “heuristic search”. The “expert systems” paradigm narrowed the focus from general intelligence to human experts in specific domains such as chemistry and medicine, and attempted to replicate their knowledge and decision-making processes. This led to the first major medical AI system, MYCIN, and eventually to more familiar software such as TurboTax.
While these produced some practical results, although limited, both of these AI paradigms failed to produce the “thinking machines” they had promised. The current “machine learning” (ML) paradigm has overcome some of the barriers to real-world relevance, thanks to a growing abundance of human-generated data, massive increases in computing power, and the revival of neural networks and other machine learning algorithms. These “learning” algorithms can be “trained” to extrapolate patterns from human-generated data, and thus, do not require programmers to explicitly represent knowledge (Garvey, 2018).
AI and computational algorithms call for rethinking the drug discovery development processes while being mindful of their prospects and challenges. They can be harnessed, however, in light of exciting omics expertise so as take into account a variety of molecular variation among individuals and populations, which may be indicative of treatment response or side effects, and thus guide drug discovery and development pipelines.
These features often include genomic variant types, such as point mutations, deletions, insertions, and translocations of gene sequences, which may be direct molecular targets for the development of drug therapies. Such examples include the clinically actionable alterations, which were found within the EGFR and ALK genes and which may be targeted with kinase inhibitor drugs (Vogelstein et al., 2013). Clinical information about the different types of genetic variants can be extracted from a couple of databases, such as ClinVar (www.ncbi.nlm.nih.gov/clinvar/), COSMIC (https://cancer.sanger.ac.uk/cosmic), and OMIM (www.omim.org).
Despite the rapid development of in silico prediction algorithms, the computational prediction of drug responses, especially in complex and multifactorial diseases, remains a challenge. The big volume and the heterogeneity of the data often inhibit the improvement of the performance of computational prediction models. Critical questions in their development often include choosing the appropriate data sets for training and testing models, selecting the most suitable computational approaches for application, as well as validating and evaluating these computational models.
Combining genomic information from next-generation sequencing approaches of thousands of diseased individuals with clinical information of disease characteristic traits and treatment outcomes may potentially lead to the identification of treatment response-associated markers through a multivariate modeling procedure. To this end, supervised machine learning algorithms enable multimarker prediction of drug responses by implementing multiomics and multitask learning approaches, which extract information across patient samples as well as across drug similarities (Azuaje, 2017).
In this study, we describe the most commonly used computational techniques, which make use of “big-scale” -omics data in most cases. We also provide details about machine learning approaches used in the drug development and drug screening process. Last but not the least, we provide a description of the available machine learning approaches for drug repositioning (also known as drug repurposing).
Overview of the Strategy of Designing Computational Prediction Models
In most cases, the development of computational prediction models for drug responses is based upon four different steps. In the first step, the appropriate datasets are selected and preprocessed first by selecting the relevant data subsets and then by normalizing them and filtering noisy or irrelevant data features (Fig. 1) (Libbrecht and Noble, 2015). These datasets may consist of single-nucleotide polymorphisms, gene copy numbers, and gene expression data. Interestingly, comparative analysis studies have shown that gene expression data have the most accurate predictive accuracy, while integrated models increased, on a marginal level, the drug response prediction accuracy (Costello et al., 2014).
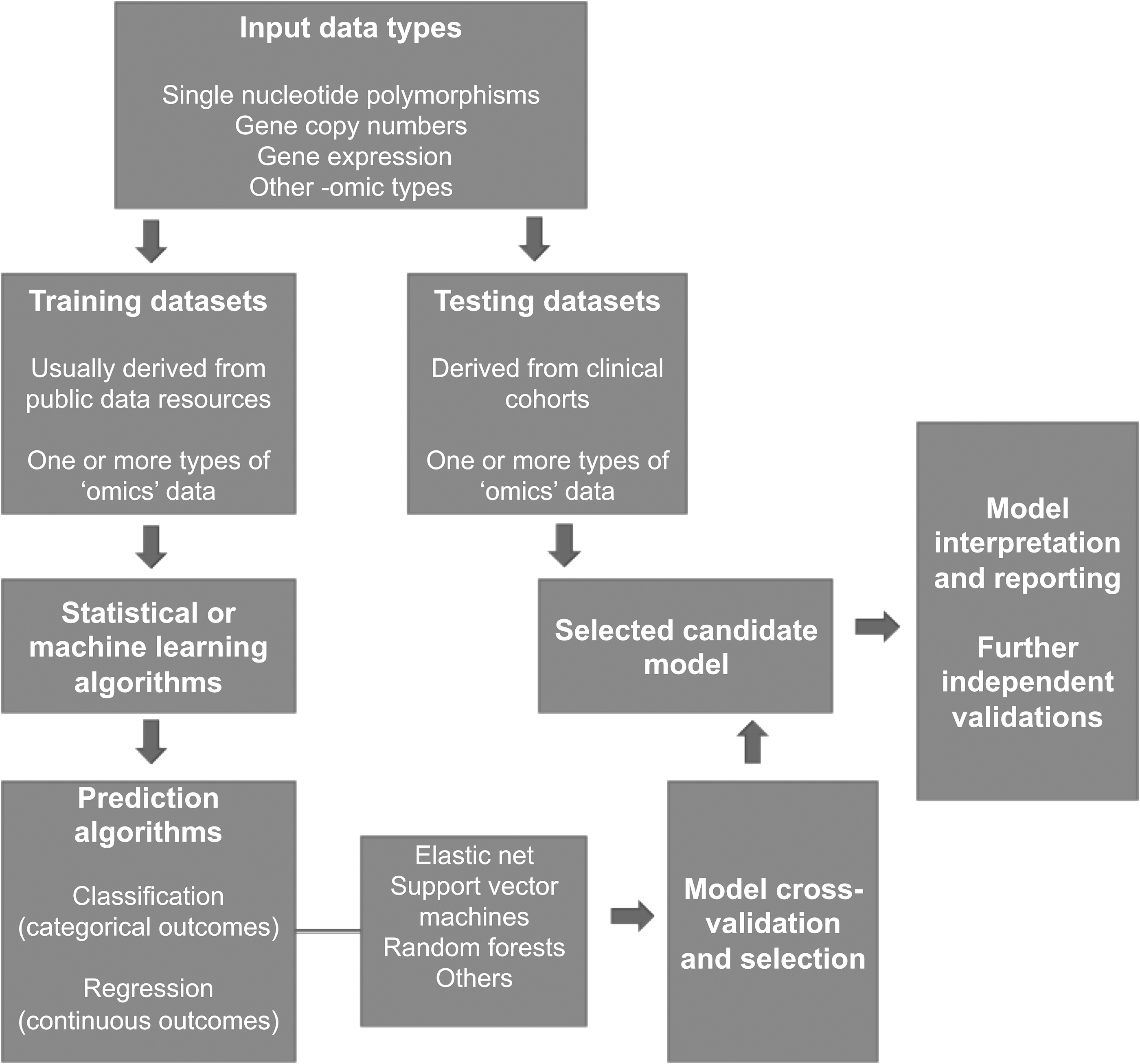
Schematic diagram illustrating the key steps in the development of computational models for predicting drug response. Data sets are obtained from data resources and they are subsequently used as training data sets, since they usually contain one or more types of “omics” data, for example, transcriptomics and DNA sequence. Such data are used as inputs to statistical or machine learning techniques. The prediction algorithms may be categorized as classification or regression models depending on the expected prediction outcomes. Then, the predictive performance of the model is assessed with cross-validation sampling techniques and the selected candidate model is further validated and tested in independent public or private datasets.
The possibility of having “extreme” responses to treatments needs to be taken into account when implementing algorithms to estimate drug sensitivity. To this end, recent models incorporate a much wider range of cell lines and/or clinical samples for a more accurate estimation of the tumor heterogeneity and the treatment responses (Azuaje, 2017).
The second step of the algorithm design includes the training phase of the model selected to address the prediction problem (Fig. 1). A variety of machine learning techniques can be implemented and the data can be fit into different models to allow a better estimation of the predictive quality of the derived models. The type of input data and the specific characteristic traits of the drug response prediction problem require careful consideration. The third step of designing the computational prediction model is often referred to as independent evaluation. During this step, multiple tests are conducted on independent data. This aims to verify that the candidate prediction model can accurately predict drug responses on unseen data derived from different laboratories and measurement platforms.
The final step includes the application of the model to data that resemble in a clinical level the characteristics of the cancer type under investigation. For example, models trained on data derived from cell lines may be tested on solid or liquid biopsies from patients. Again, the interpretation and the implementation of the appropriate methods for reporting predictions are crucial parameters (Fig. 1).
Two specific steps, which should be taken into consideration when designing the in silico prediction model, are feature selection and model evaluation. These important steps are described in detail below.
Feature Selection
Determining the most useful information-wise variables is an important step in the machine learning procedure, since it significantly affects the derived models. For this purpose, both general purpose algorithms (e.g., elastic net regression or random forest) as well as algorithms adjusted to the specific purpose of use (e.g., algorithms that consider a drug's mechanism of action) can be used (Ammad-Ud-Din et al., 2017; Yang et al., 2018).
Model Evaluation
Model evaluation is a critical and necessary step when building a robust and accurate prediction model, since the model should perform satisfactory on unseen data. Model evaluation is performed after fitting the algorithm to the training dataset and by using independent validation datasets to examine the final model. Among the most important evaluation metrics for classification applications are the overall model accuracy, precision, recall, the area under curve, as well as the precision-recall curves. Similarly, the most common metrics are correlations for regression-based models, are the root-mean-squared errors, and the coefficient of determination (Baldi et al., 2000; Berrar and Flach, 2012).
The usual process of evaluation of the fitted model is known as cross-validation (CV). During this process, the initial dataset is separated into two distinct datasets, of which one will be used for fitting the algorithm and the other one for testing how the model performs. The most commonly applied cross-validation schemes are the K-fold (KF-CV) and the leave-one-out (LOO-CV) cross-validation methods. During KF-CV, the initial dataset is divided into K-parts, with K-1 parts used for training and the remaining one used for testing. In LOO-CV, a single sample from the initial dataset comprises the left-out dataset and is left as testing. Then, the procedure is repeated as many times as the data points (Baek et al., 2009; Varma and Simon, 2006).
Public “-Omics” Data Resources for Building Drug Prediction Algorithms
As explained in the previous section, the majority of drug response prediction models are trained on data sets generated by different research consortia. Although this approach may harbor biological accuracy, it is characterized by a number of limitations, such as the number of patients analyzed or the false reads in the “-omics” and clinical data that could be potentially incorporated into model development. An alternative approach, which is gaining significant support from the community, is the training (and/or testing) of models based on publicly available data generated by large research consortia (Table 1).
Summary Details of the Key “-omics” Datasets Publicly Available from Representative Cancer Cell Lines and Patient Genomic Resources, Alongside with the Cancer Type and the Number of Samples for Each Dataset
TCGA, the cancer genome atlas; GDSC, genomics of drug sensitivity in cancer.
In a collaborative effort toward improving precision oncology, the DREAM7 (Dialogue on Reverse Engineering Assessment and Methods) Challenge co-organized together with the National Cancer Institute (NCI) analyzed the prediction accuracy of a total of 44 drug sensitivity prediction algorithms(Costello et al., 2014). Moreover, the NCI-DREAM7 challenge analyzed the genome-wide -omics profiles of 53 human breast cancer cell lines to train the prediction algorithms (Table 1). It was observed that the variability in the drug response levels across the different cell lines could be potentially explained by the genome-wide expression data, since the other -omics profiles only partially improve the performance of the prediction algorithm (Jang et al., 2014).
In another attempt toward improving the prediction accuracy of the drug sensitivity algorithms, the contribution of proteomics profiling was tested on NCI-60 pan-cancer cell line data (Ali et al., 2018; Shoemaker, 2006). The NCI-60 cell line panel includes 60 cell lines from nine different cancer types, while ∼15,000 anticancer drug treatments are tested on this panel (https://dtp.cancer.gov/discovery_development/nci-60/) (Table 1). More precisely, Ali et al. (2018) implemented an integrated Bayesian efficient multiple kernel learning model and a variety of multiomics profiles (i.e., mass-spectrometry [MS]-based proteomic profiling and target reverse phase protein array profiles) to predict drug response. This proposed model improved significantly the drug sensitivity prediction performance compared to the prediction outputs as produced only from gene expression data (Ali et al., 2018).
Of note, genomic and molecular profiling has been performed not only in cancer cell line panels but also in patient tumor samples. The Cancer Genome Atlas (TCGA) program is a landmark cancer genomics program, which provides a comprehensive cohort of -omics and clinical information across 33 different cancer types and integrates the genomic, molecular, proteomic, and clinical features from more than 11,000 cancer patients (Table 1). Geeleher et al. (2017) proposed a methodology that allowed the use of clinical cancer sequencing datasets, such as the TCGA data, for the identification of pharmacogenomics biomarkers without requiring the collection of drug responses from the patients (Geeleher et al., 2017).
A machine learning-based approach was implemented, in line with the one described by Geeleher et al. (2014), in which gene expression-based predictive models of drug response were constructed from nearly 1000 cancer cell lines from Genomics of Drug Sensitivity in Cancer (GDSC) project. These predictive models were then applied to gene expression data in the 10,000 TCGA tumor samples, thus providing an “imputed drug response profile” for each TCGA sample over 138 drugs.
Among the publicly available data resources for investigating drug responses is the Cancer Cell Line Encyclopedia (CCLE), which offers baseline data (i.e., derived from untreated samples) obtained from different “-omics” modalities and diverse measurements of drug sensitivity in cancer cell lines (Table 1). CCLE contains mutation, gene copy numbers, and gene expression data from more than 1000 cell lines from 36 tumor sites. Moreover, drug sensitivity data from more than 11,000 experiments that tested 24 anticancer drugs on ∼500 cell lines are also publicly available. In this direction, Qin et al. (2017) created an interface for the quick identification of potential genes/targets associated with drug responses in specific cancer types. This interface was based on the genomic and pharmacologic data of cancer cell lines in the CCLE and GDSC, and it allowed CV of the results produced by each data resource (Qin et al., 2017).
Categorization of ML Algorithms Used in Drug Response Prediction Models
When it comes to drug response prediction through machine learning algorithms and pipelines, the main challenges that researchers have to face can be divided into three broader categories. The first two categories attempt to estimate the response toward a single compound, either by not taking into account the specific type of cancer (Pan-cancer single-drug response prediction models) (Jang et al., 2014; Wan and Pal, 2014) or by focusing on a specific type of cancer or drug (Drug/Cancer-focused response prediction models) (Chen et al., 2015; Tran et al., 2014). Moreover, attempts are also made to compute the effects of drug combinations (Combinatorial drug response prediction models), since they are frequently used in clinical settings (Bansal et al., 2014; Yang et al., 2015).
Characteristic examples are the models based on the DREAM Challenges initiative, as mentioned above, with DIGRE being the one to stand out (Table 2). Its main hypothesis is that the effect on gene expression levels induced by the first drug under investigation contributes to those of the second. To sum up, the algorithm takes into account the information regarding the differential expression due to each compound separately to infer conclusions on their combined action (Bansal et al., 2014).
Summary of Selected Available Drug Response Prediction Algorithms and Their Key Features, Alongside with a Summary of Some of the Characteristic Models Used for Feature Selection
Classic ML Approaches in Drug Response Prediction
In general, machine learning techniques are categorized into the supervised ones, which take into account information about the classes of the training data, and to the unsupervised ones, which aim to create groups within the training data. Regarding the latter ones, the data points within each group resemble with each other as much as possible, while these data points resemble as less as possible with points belonging to other groups. Usually, analysis based on unsupervised methods is preceded and can be used to get a first glance at the data (Byers et al., 2013; Gholami et al., 2013; Nicolau et al., 2011).
When it comes to inferring anticancer drug response, unsupervised algorithms, such as k-nearest neighbors (k-nn), are widely used to create clusters of genomic and/or molecular profiles and then identify medications that exhibit differential effectiveness in some clusters (Hoadley et al., 2014). Alternatively, drug-response outcomes can be grouped together and then possible relationships with specific genomic or molecular features may be investigated (Andersson et al., 2017; Frismantas et al., 2017; Pemovska et al., 2013; Tyner et al., 2013).
Supervised methods encompass a wide spectrum of techniques, which is divided into two main categories: the main one is classification and regression-based algorithms with the first being used to determine the class of a new entry, for example, whether a cancer cell line is expected to respond in a good or unwanted way after the administration of a specific drug (Fersini et al., 2014; Jang et al., 2014), and the second one is estimation of the value of a variable in question (Falgreen et al., 2015; Neto et al., 2014). Among the most widely used algorithms for classification are Support Vector Machines and Random Forests (Amin et al., 2014; Chen et al., 2015; Cortés-Ciriano et al., 2015; Stetson et al., 2014; Tran et al., 2014) (Table 2). However, improved predictive performance under a variety of conditions (Jang et al., 2014) may be achieved by using models based on regression, such as elastic net or ridge regression (Consortium, 2015).
Improving the Performance of Prediction Models
Alternative approaches, which aim to improve predictive potential, might focus on the way that the information is represented and incorporated in the input (Consortium, 2015; Daemen et al., 2013; Neto et al., 2014). Alternatively, they might incorporate ensemble models (Cortés-Ciriano et al., 2015; Wan and Pal, 2014) or even variations of classic algorithms and frameworks, similar to those represented by the STREAM algorithm (Neto et al., 2014), which incorporate the Bayesian inference with ridge-regression or combine the net regression with Principal Components Analysis (Park et al., 2014) (Table 2).
An example worth discussing is that of TANDEM algorithm (Aben et al., 2016) (Table 2). The researchers, after considering the fact that transcriptomic data seem to be the determinant feature of drug response, attempted to explain the outcomes by utilizing all the other available information (e.g., genetic mutations, epigenetic data and so on). Gene expression was initially excluded and used only for interpretation of the remaining variability. Additional models that rely on integrating supplementary information in the training datasets are component-wise Multiple Kernel Learning (cwKBMF) and Transfer Learning (Table 2).
cwKBMF allows further characterization of specific subsets of the original data (e.g., incorporating biological pathways), which can be linked with a drug's mechanism of action (Ammad-Ud-Din et al., 2016; Yang et al., 2018). Transfer Learning was initially presented by Turki et al. (2018) and used gene expression data and tissue-specific responses during its learning phase (Turki et al., 2018). Then, data about related tissue-types were incorporated in the algorithm, while assessing how the additional data will be aligned with the ones used for training (Wang and Mahadevan, 2008).
Proposing Alternative Computational Approaches for Improving Drug Response Prediction Accuracy
Vougas et al. (2019) recently proposed an in silico mining method for gene-drug selection and drug response prediction by extracting molecular profiling information from both public and private data resources (Vougas et al., 2019). This in silico screening pipeline identifies genes as candidate drivers of drug response. Moreover, it explores large sample-spaces, while detecting low-frequency events and evaluating statistical significance even in the multidimensional space. The results are also presented in the form of interpretable and easily understandable rules. Another important feature of this novel data mining process is its ability to investigate meaningful one-way and complex relationships, providing this way complementarity to existing frameworks.
Previous studies, which also focused on identifying gene targets for drug response, implemented either the elastic net regression method or the random forests to highlight associations among multiple genes and transcripts and identify response signatures for the tested drugs. Agrawal et al. (1993) initially described the Apriori algorithm, which allows extraction of significant associations from all of the possible feature combinations from the main input dataset (i.e., tissue of origin, gene expression, mutation status, gene copy number variation, and drug response). This step is usually followed by generation of a large-rule set that contains all possible gene-to-drug and tissue-to-drug associations.
It is highly recommended that the potential biological relevance of any derived significant association rules shall be examined both computationally (based on prior knowledge) and experimentally. After comparing the derived association rules with previous frameworks, the degrees of overlap indicate the necessity of applying multiple analytical techniques to achieve the maximal information retrieval.
Another study using the Apriori algorithm is by Kuo et al. (2009), where the suitability of the Apriori algorithm was tested for the detection of adverse drug reactions (ADR) in health care data. More precisely, Kuo et al. used the Apriori algorithm to perform association analysis on the characteristics of patients, the drug treatment, their primary diagnosis, any comorbid conditions, and the ADRs or adverse events (AE), which they may experience. This analysis produced association rules indicative of the treatment combinations and the patient characteristics leading to ADRs.
During the last decade, deep learning has also emerged as a “key player” in the field of machine learning. The more advanced form of deep learning is deep learning neural networks (DLNN), which have demonstrated improved performance (i.e., classification accuracy) in various AI research areas, such as image and voice recognition or natural language processing, compared to classical computational methods.
Deep learning can be described as a class of machine learning algorithms, which uses multilayers to extract higher level features from raw input data. As the “neural” part of their name indicates, neural networks consist of input and output layers, as well as hidden layers that transform the input into information that the output layer can use (Fig. 2). Overall, DLNN is based on the modeling of high-level neural networks into flexible, multilayer systems of connected and interacting neurons that perform numerous data abstractions and transformations (LeCun et al., 2015).
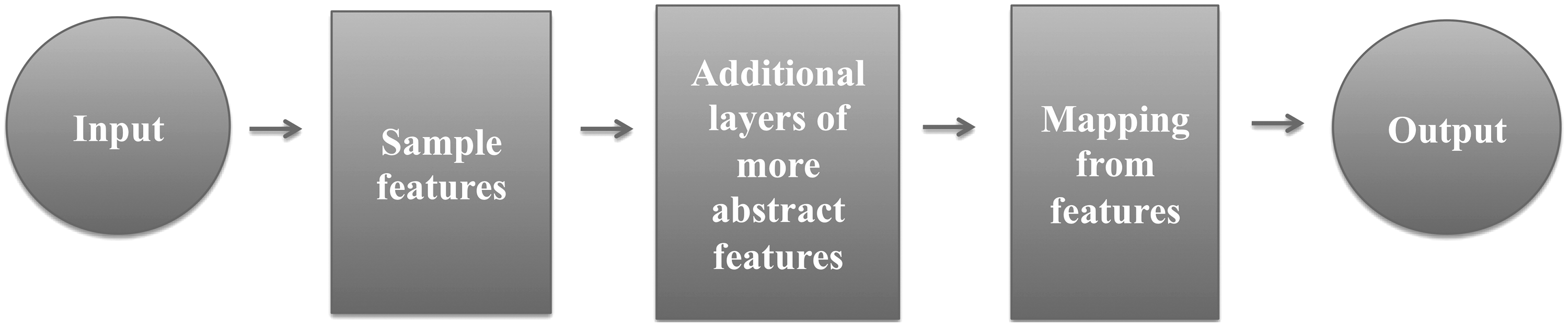
Schematic diagram illustrating the primary principle behind the deep learning neural networks.
Applications of deep learning in pharmaceutical research are ever increasing and go beyond bioactivity predictions, while showing promise in addressing diverse problems in drug discovery. Deep learning algorithm examples in pharmaceutics cover areas from bioactivity prediction models to ligand–protein interactions and from de novo molecular design to synthesis prediction models (Chen et al., 2018).
Machine Learning Approaches for Drug Repositioning
Drug repositioning (or drug repurposing) can be defined as the process of selecting a known drug for an alternative pharmacological use. This process comprises only four steps, which are as follows: compound identification; compound acquisition; development; and FDA postmarket safety monitoring. Owing to the fast growth of bioinformatics knowledge and large-scale “-omics” data, drug repositioning has decreased significantly the time cost for the drug development process. Nowadays, researchers only need ∼1–2 years to identify new potential drug targets and about 8 years to approximately develop a repositioned drug (Fig. 3).

Schematic diagram illustrating the four main steps behind the drug repositioning process.
Among the most well-known tools and platforms, which are used in developing pipelines of repositioned drug candidates in a variety of diseases, are Biovista (www.biovista.com/) and Drug Repurposing (https://drugrepurposing.info/). For example, Biovista is supported by a platform called COSS™ (Clinical Outcomes Search Space), which assists scientists in uncovering nonobvious correlations between drugs, molecular targets and pathways, as well as AEs and diseases, while simultaneously constructing evidence-based biological plausibility rationale on a systematic and highly predictable basis.
The majority of the existing computational approaches used in drug repositioning are based on gene expression response data of different cell lines after drug treatment. Alternatively, the drug repositioning approaches can be based on the combination of different types of data about disease–drug relationships, which can then be divided into several types from different viewpoints (Gonen, 2012; Lotfi Shahreza et al., 2018; Napolitano et al., 2013; Zou et al., 2013). More specifically, drug repositioning methods can be grouped according to the biological networks used (Lotfi Shahreza et al., 2018) or they can be divided into two types: data-driven and hypothesis-driven (Gonen, 2012). In summary, drug repositioning approaches can be divided into three categories: network-based approaches, text-mining approaches, and semantic approaches.
Network-based methods can be further divided into the clustering and the propagation approaches. The former ones search for relationships between drugs and diseases or putative targets into smaller clusters within a bigger network, while the latter ones are based on the flow of previously acquired knowledge through the different layers of a network. Moreover, network-based propagation approaches appear to be quite effective in identifying the interconnections of interest (Emig et al., 2013).
When examining propagation-based methods according to the way they treat the network to extract information, two main categories are arising. The first includes approaches that focus on sections of a network (local approaches), while approaches of the second category examine the network in its entirety (global approaches) (Emig et al., 2013).
Finally, a distinction can be made regarding the composition of the networks created. More explicitly, those formed using one type of information, such as Protein-to-Protein Interactions are characterized as homogeneous, while networks incorporating various data types, such as those deriving from “–omics” assays are known as heterogeneous (Wu et al., 2013).
In contrast, the text mining approach is exploiting the vast amount of available literature, which is subsequently filtered to retain only the relevant sources to extract knowledge for a set of biological terms of interest. When it comes to drug repositioning, the reference method is described as the “ABC” method. Interestingly, a series of approaches, which aim to identify relationships between drugs and diseases, is based on the “ABC” method (Weeber et al., 2001). The main idea is that when concepts A, B, and C are examined, where concept A is related to B and B to C, then it is possible that a connection exists between A and C also (Weeber et al., 2001). Finally, in semantics-based approaches, the creation of networks is guided from prior biomedical knowledge extracted from databases and then machine learning algorithms are utilized to identify new interactions and relationships, which are present in these networks (Xue et al., 2018).
Moreover, a worth mentioning application in drug repurposing is the one proposed by Zhao and So (2017), where transcriptomics data are used as the sole determinant of a drugs' potential to be characterized as a candidate drug for receiving a new indication, while focusing only on psychiatric conditions (Zhao and So, 2017). According to this study, a number of supervised machine learning algorithms were tested yielding similar results, while the presented framework can be easily adapted to any other condition of interest and set of drugs as long as there are measurable differential expression levels after drug treatment. The approach was validated through enrichment analysis of clinical trials for the compounds suggested as candidates, with the results supporting its utility.
Discussion and Future Outlook
In this article, we described some common examples of ML approaches (i.e., decision trees, random forests, LASSO) as well as alternative computational approaches (i.e., deep learning, Apriori algorithm), which can be used at different stages of drug discovery and/or drug repositioning.
Nowadays, all stages of drug discovery and development from target selection and validation to clinical trials can be subjected to and benefitted from the application of ML algorithms and pipelines. Among the different tasks that can be tackled is the challenging process of identification of novel molecular targets and the acquisition of a deeper insight of both disease mechanisms as well as complex and multifactorial disease phenotypes (Ferrero et al., 2017; Godinez et al., 2017).
AI approaches can be of great help in essentially every step of the rigorous drug design and development process. Starting from screening of vast compound libraries, hit discovery, and lead identification (Anderson, 2012; Zhu et al., 2013) to exploring the possible modifications that lead to compounds with improved pharmacokinetic, pharmacodynamic, and toxicological characteristics, planning the synthetic route to follow is usually through retrosynthesis (Mak and Pichika, 2019; Vamathevan et al., 2019). ML algorithms can also be utilized toward identifying new biomarkers for improving the drug efficacy, thus boosting the field of precision medicine (Mamoshina et al., 2018).
Moreover, recently developed ML-based biomarker discovery and drug sensitivity prediction models are promising to significantly improve clinical success rates, through contributing to the uncovering of molecular mechanisms of actions and offering the knowledge needed to personalize treatment strategies (Kraus, 2018; Li et al., 2015). This is primarily achieved through the analysis, exploration, and interpretation of -omics data via AI and machine learning approaches. Further implementation of these ML-based pipelines in other fields of the pharmaceutical industry includes chemoinformatics, computational genomics, and biomedical imaging (Angermueller et al., 2016; Ekins, 2016; Gonczarek et al., 2018).
It is worth to note that alternative approaches include the integration of systems biology perspective with AI and ML techniques for drug development and drug repositioning. These alternative approaches can be signature-based or network-based. Signature-based approaches exploit gene expression motifs between drug and disease phenotypes, while investigating for new drug-disease associations. Network-based approaches implement the “guilt-by-association” principle, thus leading to discovery of hidden drug-disease associations through knowledge-based or computationally driven networks (Park, 2019). These networks could be either evidence-based relying primarily on experimental evidence or statistically inferred networks relying on the derived components from statistical analysis (Greene and Voight, 2016).
Implementation of the systems biology perspective via ML approaches lies so far primarily within the field of drug repositioning. Among the most successful examples, where systems biology and ML have boosted drug discovery, are the uncovering of the Proprotein convertase subtilisin/kexin type 9 (PCSK9) association with heart disease (Cohen et al., 2005, 2006) as well as the association of missense (Sladek et al., 2007) and loss of function (Flannick et al., 2014) mutations within islet-specific zinc transport gene (SLC30A8) with a risk or protective role against diabetes, respectively.
The introduction of ML and deep learning algorithms and pipelines was enabled by the increased computational power combined with the availability of “big data”, with various pharmaceutical companies recently investing in it. However, despite the high expectations regarding ML-approaches in drug discovery and precision medicine, there are only a few cases where biomarkers and predictive models have been implemented in clinical trials. Important factors, affecting the adoption of such predictive models, are the model selection and reproducibility of models build using neural networks, the access to curated data as well as the design of assays suitable for a clinical setting (Vamathevan et al., 2019).
Among the most important limitations that need to be carefully considered in the field of ML in drug discovery and drug repositioning is the quality of the experimental data. A model can be only as accurate as the training data. If the data are flawed, contain noise, and their collection and integration do not follow a systematic procedure, then it is most likely that the algorithm prediction will not be accurate. If the algorithm and its users do not take those biases into account, then the ML prediction outputs will also be flawed (Vamathevan et al., 2019).
Taken together, machine learning and deep learning methods remain a ‘black box’ (Lamberti et al., 2019). Since the model features are not explicitly specified, the creator of the algorithm may be unaware of what is being inspected during the intermediate stages, or the exact process leading to the specific outcome/prediction of the algorithm. To this end, recruitment of the appropriately trained staff to bridge the gap between -omics and AI, is another issue that should be carefully considered.
Regardless of the current limitations, the partnership between pharmaceutical companies and companies with a focus on developing ML and deep learning approaches could help toward identification of novel small molecules, discovery of new treatment methods, or toward monitoring the massive health data via applicable technologies. Given the high cost, time-, resources-, and effort-wise, of designing and developing new medications, which is accompanied by decreased approval rates, it is reasonable to rely on computational approaches aiming to further improve the effectiveness of drug development while reducing the failure percentage. These advances will act as contributing factors to the improvement of health care services, precision medicine, and drug efficacy (Mak and Pichika, 2019).
Conclusions
In this study, we analyzed and described several different machine learning algorithms that can be used for in silico drug screening, while giving examples of alternative approaches for this purpose. These algorithms, offering a wide spectrum of exploration capabilities, allow the identification of biomolecules, the inhibition or enhancement of which may improve the effectiveness of therapeutical approaches. Moreover, when data-mining scheme are implemented, then meaningful gene-to-drug response relationships can be captured. To this end, we also provide a summary of useful ML approaches utilized in drug repurposing, which is an alternative approach to the traditional drug discovery approach. To conclude, the current review shows that the field of AI, encompassing machine learning, as well as approaches such as deep learning can be implemented in clinical practice toward the improvement of precision medicine and drug discovery/drug screening process.
Footnotes
Acknowledgments
All authors have met the ICMJE criteria for authorship. GPP is Full Member and National Representative at the European Medicines Agency, Committee for Human Medicinal Products (CHMP) - Pharmacogenomics Working Party in Amsterdam, the Netherlands. The views expressed are the personal opinions of the authors only.
Author Disclosure Statement
The authors declare they have no competing financial interests.
Funding Information
No funding was received in support of this article.