
Abstract
Artificial intelligence, machine learning, health care robots, and algorithms for clinical decision-making are currently being sought after in diverse fields of clinical medicine and bioengineering. The field of personalized medicine stands to benefit from new technologies so as to harness the omics big data, for example, to individualize and accelerate cancer diagnostics and therapeutics in particular. In this overarching context, breast cancer is one of the most common malignancies worldwide with multiple underlying molecular etiologies and each subtype displaying diverse clinical outcomes. Disease stratification for breast cancer is, therefore, vital to its effective and individualized clinical care. The support vector machine (SVM) is a rising machine learning approach that offers robust classification of high-dimensional big data into small numbers of data points (support vectors), achieving differentiation of subgroups in a short amount of time. Considering the rapid timelines required for both diagnosis and treatment of most aggressive cancers, this new machine learning technique has important clinical and public applications and implications for high-throughput data analysis and contextualization. This expert review describes and examines, first, the SVM models employed to forecast breast cancer subtypes using diverse systems science data, including transcriptomics, epigenetics, proteomics, and radiomics, as well as biological pathway, clinical, pathological, and biochemical data. Then, we compare the performance of the present SVM and other diagnostic and therapeutic prediction models across the data types. We conclude by emphasizing that data integration is a critical bottleneck in systems science, cancer research and development, and health care innovation and that SVM and machine learning approaches offer new solutions and ways forward in biomedical, bioengineering, and clinical applications.
Introduction
Diagnosis and treatment of cancers remain the most formidable challenges in health care innovation. A critical bottleneck in this context is data integration and sense-making. The recent rise of artificial intelligence, machine learning, and other high-throughput data analysis algorithms offers new prospects. In this context, the support vector machine (SVM) is a supervised machine learning algorithm for classifying data sets (Schölkopf et al., 2013). SVM has been reported to display higher performance compared with the other traditional supervised classifiers such as decision tree (Rokach and Maimon, 2005) and supervised artificial neural network (Zhang, 2016). SVM classifies high-dimensional big data into small numbers of data points (support vectors), thus achieving differentiation of subgroups in a short amount of time.
Considering the rapid timelines required for both diagnosis and treatment of most aggressive cancers, this new machine learning technique has important clinical and public applications and implications for high-throughput data analysis and contextualization.
This expert review describes and examines, first, the SVM models employed to forecast breast cancer subtypes (BCSs) using diverse systems science data, including transcriptomics, epigenetics, proteomics, and radiomics, as well as biological pathway, clinical, pathological, and biochemical data. Then, we compare the performance of the present SVM and other diagnostic and therapeutic prediction models across the data types.
Support Vector Machines
Relevance to personalized medicine and cancer research
In the SVM algorithm, a decision boundary, forming two sides of different classes, is drawn to separate the data points. This imaginary boundary is called hyperplane. During classification, data points belonging to these different sides of the hyperplane are optimized in a way to provide the highest distance margin between two sides of data. The data points at the acceptable proximity to the boundary are called support vectors. They are used for the coordinate optimization of the decision boundary.
This technique is important for its application in classification and selection of informative features, such as genes, proteins, and pathways. Yet, the classification of big data is still computationally expensive. Thus, in various applications, a hybrid methodology of the SVM, including a data filtration component, is observed. Besides, high dimensionality (multiple parameters) in applied data sets has no negative effect over SVM. Therefore, groups having small sample sets and a high number of dimensions can be properly classified (Gao et al., 2017).
Breast cancer is the most frequent malignant disease among women worldwide and is also the leading cause of cancer mortality after lung cancer (Bray et al., 2018). Several clinical and pathological features such as tumor size and grade, lymphovascular invasion of tumor cells, and axillary lymph node (ALN) status are reported as important factors in the determination of breast cancer besides the presence of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor-2 (HER2) on the surface of the tumor cell and the expression level of Ki-67 (Coates et al., 2015; Yamanouchi et al., 2019). Breast cancer is also known for having heterogeneity under both spatial and temporal dimensions, causing problems during the dissection process (Agner et al., 2014; De Ronde et al., 2014; Hsu et al., 2012; Tyanova et al., 2016; Vidić et al., 2018; Yersal and Barutca, 2014).
According to the current classification system (Goldhirsch et al., 2013), BCSs are defined as luminal A (ER and/or PR positive, HER2 negative, low levels of the protein Ki-67), luminal B (ER and/or PR positive, high levels of Ki-67) with HER2 negative, luminal B with HER2 positive, HER2 enriched (ER and PR negative), and basal like (triple-negative breast cancer, TNBC). It is important to identify the BCSs not only for evaluating the prognosis of the disease but also for providing adequate therapy since drug responses of subtypes show variations (Januškevičienė and Petrikaitė, 2019) (Fig. 1).
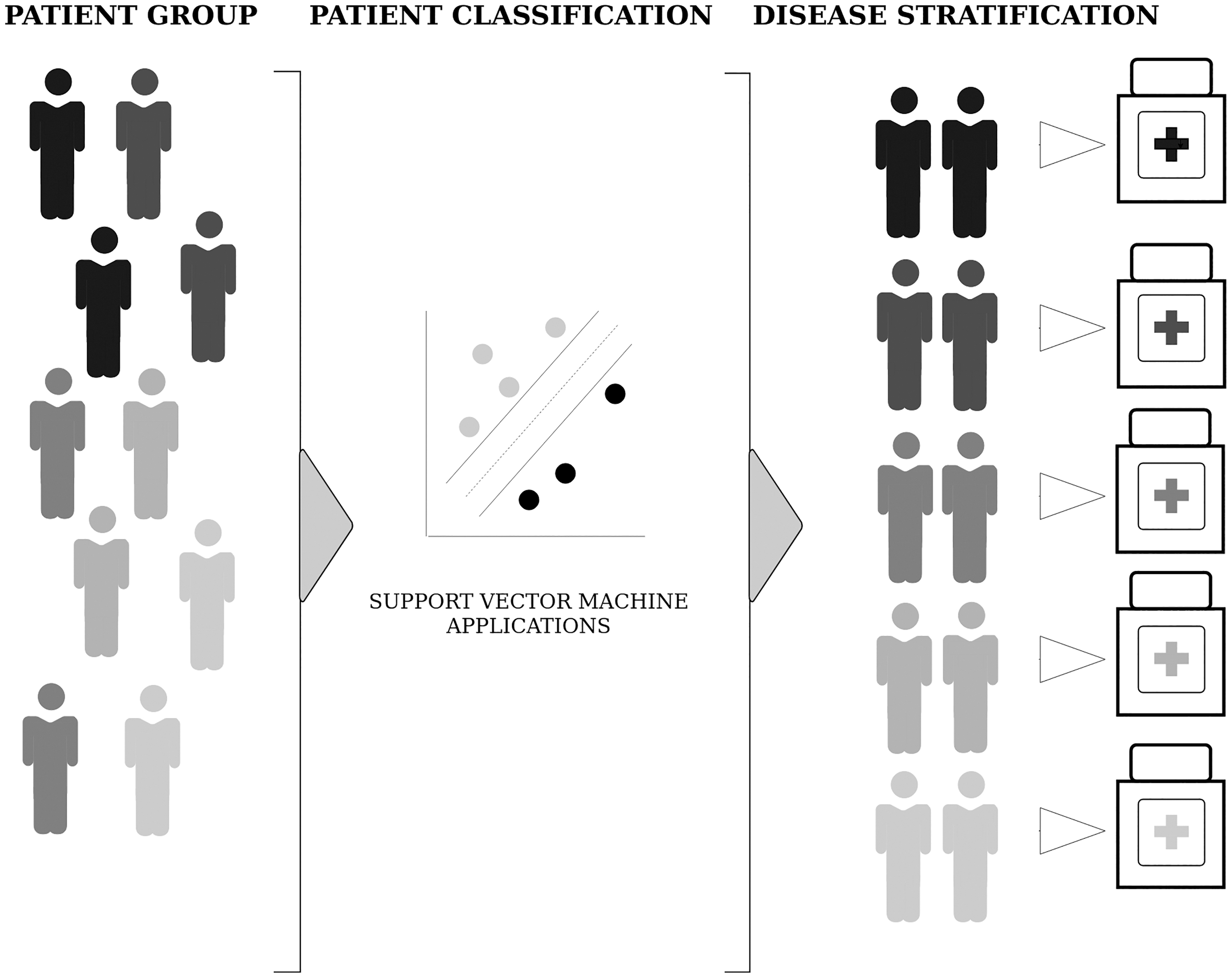
A conceptual description of disease stratification using support vector machine applications.
In this study, we presented a brief review of BCS classification through SVM models, discussed the applicability of various data types in these models, and compared the accuracy of the present prediction models. We also emphasize the applicability of SVM in disease stratification using breast cancer as a case study. It should be noted that once the problem of lack of data integration strategies is solved, the predictive performance of the models would highly improve.
Omics-Based Prediction Models
There are SVM models that were developed using x-omics data sets, which include high-throughput data from different biological levels (Table 1). Depending on the quality of data, these models present sufficient prediction accuracies.
Selected Studies with Significant Relevance to Stratification of Breast Cancer Subtypes Using Support Vector Machines
2D-DIGE, 2-dimensional, differential in-gel electrophoresis; LOOCV, leave-one-out cross-validation; miRNA, microRNA; DCE-MRI, dynamic contrast-enhanced magnetic resonance imaging; RNA-seq, RNA sequencing; SILAC, stable isotope labeling with amino acids in cell culture; SVDD, support vector data description; SVM, support vector machine; v-SVM, variant of support vector machine.
Expression levels of coding and noncoding RNAs (ncRNAs) differ in physiological conditions and specific stages of development and therefore are frequently used to construct predictive models based on intercondition expression differences of messenger RNAs (mRNAs) and ncRNAs. Similarly, mRNA and microRNA (miRNA) transcriptome data sets produced using different technologies, such as RNA sequencing (RNA-seq) or microarray, are among the most frequently used omics data sets employed with machine learning techniques, including SVM for the differentiation of BCSs (De Ronde et al., 2014; Hsu et al., 2012; Lan et al., 2018; Sokolov et al., 2016). Typically, SVM methods are applied by using two or more classes. However, contrary to two-class methods, negative and positive classes are also given together without affecting the accuracy of model performance in the one-class SVM (Sokolov et al., 2016).
Various machine learning methods employing transcriptome data sets are used in the stratification of BCSs. However, the accuracies of methods vary depending on the applied data set or the sampling used. For instance, through employment of RNA-seq data and performance comparison using the area under the curve (AUC) score, Sokolov et al. (2016) reported the outperformance of a variant of SVM (v-SVM) and logistic regression (LREG) (Dayton, 1992) compared with support vector data description (Tax and Duin, 2004). On the other hand, in another study, a similar performance comparison methodology was employed, using microarray data instead of RNA-seq, to identify subtype-specific chemotherapy response predictors.
As a result, the optimal predictors for different subtypes were identified through nearest mean (Dabney and Storey, 2007), naive Bayes (Rish, 2001), and 3-nearest neighbor (Akkus and Güvenir, 1996) along with LREG, while the SVM prediction model failed in performance (De Ronde et al., 2014). Furthermore, in the case of employing RNA-seq-based miRNA expression data in distinguishing BCSs (Hsu et al., 2012), the SVM with fivefold cross-validation indicated higher performance in terms of the AUC metric when compared with methods involving the Fisher score (Gu et al., 2012) and Hellinger distance (Lan et al., 2018; Wu and Karunamuni; 2014). High accuracy of SVM was also shown in the identification of potential miRNA biomarkers using subtype-specific microarray data.
Besides transcriptome data, genome-wide DNA methylation profiling (methylome) data were also considered in familial breast cancer to identify distinct profiles defined by mutation status. Flanagan et al. (2010) analyzed transcriptome and methylation profiles to compare the mutation status (BRCA1, BRCA2, and BRCAx) and intrinsic BCSs. The SVM with fivefold cross-validation classification by using gene expression data yielded 100% accuracy in predictions of intrinsic subtypes and 90% accuracy in prediction of the BRCA1 mutation, whereas predictions for BRCA2 and BRCAx failed. In the case of methylation profiles, intrinsic BCSs failed, while mutation studies improved (Flanagan et al., 2010). Several other studies evaluating epigenetic data through machine learning prediction models yielded a high potential in some of the classification case studies (Alag, 2019; Lo Bosco et al., 2016; Robertson, 2005).
Considering that proteomics reflects cellular functions more than information from genomics, epigenetics, and transcriptomics; breast cancer subtyping was also studied at the proteomic level (Tyanova et al., 2016; Waldemarson et al., 2016). Quantifying proteins through stable isotope labeling with amino acids in cell culture technology and SVM with the one-vs-rest approach, Tyanova et al. (2016) achieved stratification of BCSs with high accuracy. In addition, Waldemarson et al. (2016) employed two-dimensional, differential in-gel electrophoresis and microarray technologies to obtain proteomic and transcriptomic data sets, respectively. They performed pairwise comparisons among subtypes and presented a clear distinction between basal-like and luminal A tumors using SVM with the leave-one-out cross-validation (LOOCV) approach.
The predictive performance of omics-based approaches could be improved once the quality of the data is enhanced.
Pathway-Based Prediction Models
The idea that harnessing the pathway information and relationships between the cellular molecules can help reduce or eliminate the noise in omics data has paved the way for pathway-based, higher performance prediction models.
The regulatory pathways such as JAK/STAT, inflammatory mediator regulation of TRP channels, and glutamatergic synapse as well as disease pathways, including basal cell carcinoma, nonsmall cell lung cancer, and amyotrophic later sclerosis, were reported as common pathway signatures of BCSs (Karagoz et al., 2015; Turanli et al., 2019). The SVM-based classification of BCSs using pathway-based biomarkers was accurate (>90%) in the true prediction of luminal and basal-like patients, yet inaccurate in prediction of other BCSs (Wu et al., 2017).
Similarly, metastatic behavior of breast tumors was also studied using pathway-based prediction modeling, where type 1 diabetes mellitus, cytokine–cytokine receptor interaction, and Hedgehog signaling were associated with the nonmetastasis group. An overall accuracy slightly lower than 90% was achieved using SVM with 20-fold cross-validation (Graudenzi et al., 2017). These studies indicate the possible contribution of pathway-based biomarkers as features in the diagnosis and prediction of breast cancer subtyping and staging.
Imaging-Based Prediction Models
In addition to omics-based and pathway-based models, imaging-based prediction models are also frequently used. Radiomics provides quantitative imaging features enabling visualization of the phenotypes as being correlated with genetic information (Forghani et al., 2019). Acquired imaging data can be applied for development of prediction models that can provide higher characterization. In this sense, magnetic resonance imaging (MRI) and ultrasonography are widespread techniques for breast cancer diagnosis, and several studies employed the SVM process to improve the interpretation of images and to observe whether characteristics of images could provide differentiation among BCSs.
MRI data were analyzed through the SVM process with LOOCV (Sutton et al., 2016). However, the performance of the predictions was questionable and the accuracy was limited in each subtype. Similarly, analysis of high-throughput ultrasound features and information of cancer biomarkers through SVM with threefold cross-validation resulted in limited accuracy in terms of AUC values (Guo et al., 2018). On the other hand, Agner et al. (2014) showed superior performance of using computer-aided diagnosis (CAD) methods with dynamic contrast-enhanced MRI (DCE-MRI) in differentiating BCSs. Although DCE-MRI is reported as a sensitive technique in the detection of TNBC and screening BRCA mutation carriers, it was found to be problematic due to high similarity in imaging profiling of triple-negative lesions and benign fibroadenomas.
However, the employment of CAD methods has been proposed to increase diagnostic specificity of DCE-MRI since high accuracy (97%) could be achieved through SVM classification of these subgroups. In addition, integration of diffusion-derived parameters (mean, standard deviation, skewness and kurtosis of apparent diffusion coefficient, relative enhanced diffusivity, and intravoxel incoherent motion) with MRI boosted the performance of SVM significantly in the classification of benign and malignant breast tumors (Vidić et al., 2018).
Prediction Models Utilizing Clinical, Pathological, and Biochemical Data
A rich source of information on tumors can be gathered through clinical, pathological, and biochemical analyses, which should be considered as an indispensable part of cancer research especially for entailing machine learning predictions under critical and uncertain situations (Shah et al., 2019; Visweswaran et al., 2010). In this sense, a study that could be accepted as a tutorial example was presented by Wu et al. (2014), where they constructed the SVM model to classify BCSs as well as positive and negative ALN metastasis groups based on pathological information of the primary tumor and clinical features (such as age at diagnosis, tumor size, positive lymph node, histology grade, ER status, PR status, HER2 status, and the presence of lymphovascular invasion).
The SVM model correctly predicted ALN metastases in 75% of patients using pathological and clinical information. The predictive ability of BCSs using subgroup analysis showed no difference and this predictive performance was inferior, with only 60% accuracy. In the biochemical perspective, Becker-Putsche et al. (2013) constructed the SVM model using protein, lipid, and nucleic acid data acquired from Raman spectroscopy. The research was applied over six breast cancer cell lines representing different BCSs on the single-cell level. The classification performance on the cell level was observed as 97%.
Conclusions and Outlook
This expert review examined the employment of SVM prediction models in classification of BCSs by using diverse data types. The predictive performance of SVM methods involving radiomic data was significantly higher and almost without failure in discriminating BCSs. Radiomics provides big data consisting of features that are usually easy to interpret and therefore is practical considering its noninvasive feature. Radiomics and applications of machine learning approaches are likely to receive greater research and clinical interest in the near future.
In the case of consistency and cost-effectiveness considerations, omics- and pathway-based prediction models, which present acceptable accuracy, should be preferred. Thus, repeating the same or similar experimental studies could be prevented by gene expression repositories that are freely available and focusing on data-intensive computation. Despite requiring more applications for improving the consistency of data, information at the biochemical, clinical, and pathological levels could be the key feature for further classification studies.
Although these studies collectively show that biological data at various levels are indeed useful in SVM applications, it is clear that models based on the integration of diverse data will yield markedly more accurate results. Further studies should be expected to increase in this direction, and in particular, the development of new SVM models for integration of radiomic images with molecular omics data would be of great interest for health care innovation.
Disease stratification for improved cancer care and treatment is vital to realize the overarching aim of personalized medicine. SVM is a promising approach for development and application of the best medical practices and will be encountered more frequently in 2020 and the coming decade as the concept of precision/personalized medicine moves from theory to mainstream clinical practice.
We conclude by emphasizing that data integration is a critical bottleneck in systems science, cancer research and development, and health care innovation and that SVM and machine learning approaches offer new solutions and ways forward in biomedical, bioengineering, and clinical applications.
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
No funding was received for this article.