
Abstract
Mapping the normal eye proteome in healthy persons is essential to unravel the molecular basis of diseases impacting visual health. The vitreous occupies a large portion of the human eye between the lens and the retina and plays a significant role in vitreoretinal diseases as well as maintaining clarity in the visual field, providing nutrition to the lens, and protecting the eye from mechanical shocks. It comprises four distinct anatomical regions, namely the vitreous core, vitreous cortex, vitreous base, and anterior hyaloid. Among these, the vitreous is attached to other substructures in the eye by the vitreous base, which is its strongest point of attachment. Alterations in vitreous substructures have been reported in several vitreoretinal disorders, including vitreomacular traction, vitreoretinopathies, and age-related macular degeneration. There has been limited knowledge on proteomics variations at a resolution of vitreous substructures, including the functionally and pathophysiologically significant vitreous base. We report here new findings on the proteome map of the vitreous base in normal healthy tissue. We employed a global, unbiased proteomic profiling approach resulting in the identification of 6511 proteins. Of these, 302 proteins were involved in metabolic processes essential for energy utilization. Moreover, we identified several structural and nutrient transport proteins. Notably, the identified proteome repertoire indicates that the vitreous base might possess additional physiological functions and may not be a passive structure. This study constitutes the most extensive catalog of vitreous base proteins to our knowledge and offers novel insights as a baseline for future studies on the pathobiology of various eye diseases. These data also invite us to consider a potentially more active functional role for the vitreous base in eye physiology and visual health.
Introduction
The vitreous is one of the largest structures in the eye and lies adjacent to the retina posteriorly and the lens and ciliary body anteriorly. The human vitreous comprises four distinct anatomical regions, namely the vitreous core, vitreous cortex, vitreous base, and anterior hyaloid. The vitreous core is surrounded by the cortical vitreous, which comprises the anterior hyaloid membrane and the vitreous base. It is the largest compartment and houses the vitreous humor. The anterior hyaloid membrane is made up of condensation of protein fibers and is attached to the posterior lens. The vitreous base consists of dense collagen fibers, which are implanted in an area that extends 2 mm anteriorly to adjoining pars plana region of the ciliary body and 3 mm posteriorly to peripheral retina posterior to the ora serrata. The fibers also extend radially in toward the vitreous gel. The vitreous base is the strongest point of attachment of the vitreous.
The vitreous is a vital structure that presents a clear optical path for light to traverse to the retina and is also required to maintain intraocular pressure. Studies have also suggested an essential role for vitreous in maintaining the molecular and mechanistic homeostasis, including oxygen homeostasis in the eye (Foulds, 1987). The detachment of the vitreous base and anterior vitreous commonly occurs following blunt ocular trauma and maybe associated with vitreous hemorrhage, retinal hemorrhage, or dislocation of the crystalline lens (Spraul and Grossniklaus, 1997). In addition, age-related changes in the vitreous also contribute to several vitreoretinal disorders, including retinal tears, symptomatic vitreomacular traction, and retinal detachment (Sebag, 2004).
The molecular profile of various ocular tissues has been carried out using high-throughput OMICS platforms, including RNA-Seq and proteomic approaches. Importantly, the Human Eye Proteome Project (HEPP) is one of the latest initiatives of HUPO Biology and Disease-driven Human Proteome Project (B/D-HPP), which aims to provide a comprehensive of proteome data of different ocular components such as iris, ciliary body, sclera, optic nerve, tears, aqueous humor, lens, vitreous humor, retina, and retinal pigment epithelium/choroid (Ahmad et al., 2018).
Our group and others have previously cataloged the proteome expression profile of iris (Murthy et al., 2016), choroid (Dammalli et al., 2017), ciliary body (Goel et al., 2013), as well as retina (Kim et al., 2014), vitreous humor (Murthy et al., 2014), aqueous humor (Murthy et al., 2015), optic nerve (Karthikkeyan et al., 2018), sclera (Mohanty et al., 2019) and cornea (Subbannayya et al., 2020). However, in the case of vitreous substructures, only a single study has been reported to date wherein from four substructures, namely anterior hyaloid, the vitreous cortex, the vitreous core, and the vitreous base, a mean of 2062 proteins was identified from each substructure (Skeie et al., 2015).
In this study, we aimed at elucidating the proteome profile of vitreous base using high-resolution mass spectrometry. In addition to identifying several known and novel proteins in the vitreous base, we provide information on biological processes and signaling modules. For all the proteins identified, we also provide the baseline abundances. Our data provide a resource to explore further the proteins involved in vitreous-related ocular abnormalities, which will further aid in identifying therapeutic targets.
Materials and Methods
Sample details
The vitreous base tissues were dissected from three donor eyes. The tissues were made available after obtaining approval from the Institutional review board of Vittala International Institute of Ophthalmology, Bangalore, India, and adhere to the tenets of the Declaration of Helsinki. The tissues were obtained from three healthy donors and were immediately stored at −80°C until further analysis. The details of the samples are provided in Supplementary Table S1.
Sample preparation and fractionation
The snap-frozen vitreous base tissues were homogenized using mortar and pestle in liquid nitrogen. Following this, the samples were resuspended in lysis buffer containing 4% sodium dodecyl sulfate and 50 mM triethylammonium bicarbonate (TEABC), pH 7.5. The tissue lysates were sonicated using a probe sonicator, followed by heating for 10–15 min at 90°C. The lysates were cooled and centrifuged at 12,000 rpm for 10 min. The protein concentration of the cleared lysate was estimated using the bicinchoninic acid assay (BCA) kit (Thermo Scientific Pierce). Equal amounts of proteins from each of the donors were pooled for further analysis.
In-gel digestion
An equal amount of protein lysate was pooled from each sample. From this, 300 μg pooled lysate was resolved on a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel. In-gel digestion was carried out as described previously (Mohanty et al., 2019). Briefly, the gel lane was sliced into 24 bands. Each gel band was cut into 1 × 1 mm pieces, transferred to Eppendorf tubes, and destained using 40 mM ammonium bicarbonate and 40% acetonitrile (ACN). Reduction and alkylation of disulfide bonds were carried out using 5 mM dithiothreitol (DTT) at 60°C for 20 min and 20 mM iodoacetamide (IAA) at room temperature for 10 min in the dark, respectively.
Sequencing grade modified trypsin (Cat. No. V5111; Promega, Madison, WI) was added with an enzyme: substrate ratio of 1:20, and digestion was carried out at 37°C for16 h. Peptides were extracted using 80% ACN and 0.5% acetic acid, and dried. They were then reconstituted in 0.1% formic acid and desalted using C18StageTips (3 M Empore high-performance extraction disks) (Rappsilber et al., 2003) and vacuum dried. The dried peptides were stored at −80°C till liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.
In-solution trypsin digestion and high-pH RPLC-based fractionation
Protein lysates of 300 μg from pooled samples were reduced by incubating in 10 mM DTT at 60°C for 20 min. Alkylation was carried out by incubating the samples in the dark with 20 mM IAA at room temperature for 10 min. The lysate was further subjected to acetone precipitation, and the pellet was resuspended in 50 mM TEABC. Digestion was carried out at 37°C for 16 h using L-(tosylamido-2-phenyl) ethyl chloromethyl ketone treated trypsin (Worthington Biochemical Corporation, Lakewood, NJ) at a final concentration of 1:20 (w/w). The reaction was acidified using 0.1% formic acid, and the peptides were lyophilized and stored at −80°C until further use. High-pH reverse-phase chromatography was performed for the fractionation of peptides.
Briefly, the peptides were reconstituted in bRPLC solvent A (10 mM TEABC buffer, pH ∼8.5) and loaded onto a Waters XBridge column (Waters Corporation, Milford, MA; 130 Å, 5 μm, 250 × 4.6 mm) using a Hitachi LaChrom Elite High Performance Liquid Chromatography (HPLC) system. The peptide separation was achieved using a 130-min gradient at a flow rate of 0.5 mL/min of solvent A (10 mM TEABC buffer, pH ∼8.5) and B (10 mM TEABC buffer, 90% ACN, pH ∼8.5). The fractionation program employed was as follows: 97% solvent A for 20 min, followed by a 90% solvent A for 60 min, then continued by a gradient of 35–100% of solvent B for 25 min, and further subjected to equilibration in 95% of solvent A for 25 min. The fractions were collected on a 96-well plate format from the 10th min onward and were finally concatenated into 24 fractions. Pooled samples were lyophilized and stored at −80°C until LC-MS/MS analysis.
Tandem mass spectrometry analysis
The peptide digests were analyzed on Thermo Scientific Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific, Bremen, Germany), coupled to Easy-nLC-1200 (Thermo Scientific). The lyophilized peptides were reconstituted in 0.1% formic acid (Solvent A) and loaded on the trap column (75 μm × 2 cm, nanoViper, 3 μm, 100Å) at a flow rate of 4 μL/min with Solvent A and resolved on an analytical column (EASY-SPRAY RLC C182 μm 15 × 50 μm) at a flow rate of 300 nL/min.
Data were acquired by using data-dependent acquisition mode at a scan range of m/z 400–1600, in positive ion mode. The maximum injection time was set as 55 ms using an Orbitrap mass analyzer at a mass resolution of 120,000. MS/MS analysis was carried out at a scan range of m/z 110–2000, and the top 10 intense precursor ions were selected for each duty cycle and subjected to higher collision energy dissociation with 33% normalized collision energy. The fragmented ions were detected using Orbitrap mass analyzer at a resolution of 30,000 with maximum injection time of 200 ms.
Data processing
Mass spectrometry-derived data were searched against the Human RefSeq 75 protein database (consisting of 36,713 entries along with common contaminants) in Proteome Discoverer 2.1 (Thermo Scientific, Bremen, Germany) using SequestHT and Mascot (version 2.5.1; Matrix Science, London, UK) search algorithms. The parameters included trypsin as a proteolytic enzyme with a maximum of two missed cleavages. Carbamidomethylation of cysteine was specified as a fixed modification, and acetylation of protein N-terminus and oxidation of methionine were set as variable modifications with a minimum peptide length of 7 amino acids. The precursor and fragment mass tolerance were fixed as 10 ppm and 0.05 Da, respectively. The data were searched against the decoy database with a 1% false discovery rate (FDR) cutoff at the peptide level.
Bioinformatics analysis
To obtain an in-depth analysis of mass spectrometry-derived vitreous base data, the identified proteins were classified based on biological process, molecular function, and cellular component using ProteinCenter Annotation node in Proteome Discoverer 2.1. The intensity-based absolute quantification (iBAQ) method was used for calculating the relative abundance of the protein using an in-house python script. Pathway enrichment analysis was performed using the PANTHER Overrepresentation Test (Annotation version PANTHER version 11.1 Released 2016-10-24). The list of proteins identified was compared against the reference list of all human genes present in the database. Bonferroni correction for multiple testing was applied and only those pathways that were significantly enriched (p-value <0.05) were considered for further analysis.
The protein-protein interaction (PPI) and interactome analysis were carried out for the top 200 abundant vitreous base proteins using the Search Tool for the Retrieval of Interacting Genes (STRING) (www.string-db.org), where K-means clustering was used to cluster the network to three clusters. The network coordinates were fetched and exported into Cytoscape (3.7.2) (Shannon et al., 2003). NetworkAnlyzer was used to get the network parameters where the betweenness centrality values were used to plot the network (Assenov et al., 2008).
Data availability
Mass spectrometry-derived raw data were deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) through the PRIDE partner repository with the dataset identifier PXD016232 (Vizcaino et al., 2013).
Results and Discussion
Proteomic profiling of human vitreous base
An unbiased global proteomic analysis of the human vitreous base was carried out using two different fractionation strategies, namely in-gel digestion and high pH reverse-phase liquid chromatography-based separation followed by data acquisition using Orbitrap Fusion Tribrid mass spectrometer. A total of 716,629 MS/MS spectra were acquired in FT-FT (Fourier transform) mode resulting in the identification of 395,426 peptide spectral matches mapping to 38,459 peptides. Overall, these peptides corresponded to 6511 proteins, the largest reported to date. The experimental workflow employed in this study is depicted in Figure 1. Of the proteins identified in the vitreous base, we confirmed the annotated translation start site of 617 proteins based on the peptides identified with N-terminal acetylation modification. A partial list of identified proteins is provided in Table 1. A complete list of proteins and peptides identified in the human vitreous base is provided in Supplementary Tables S2 and S3, respectively.
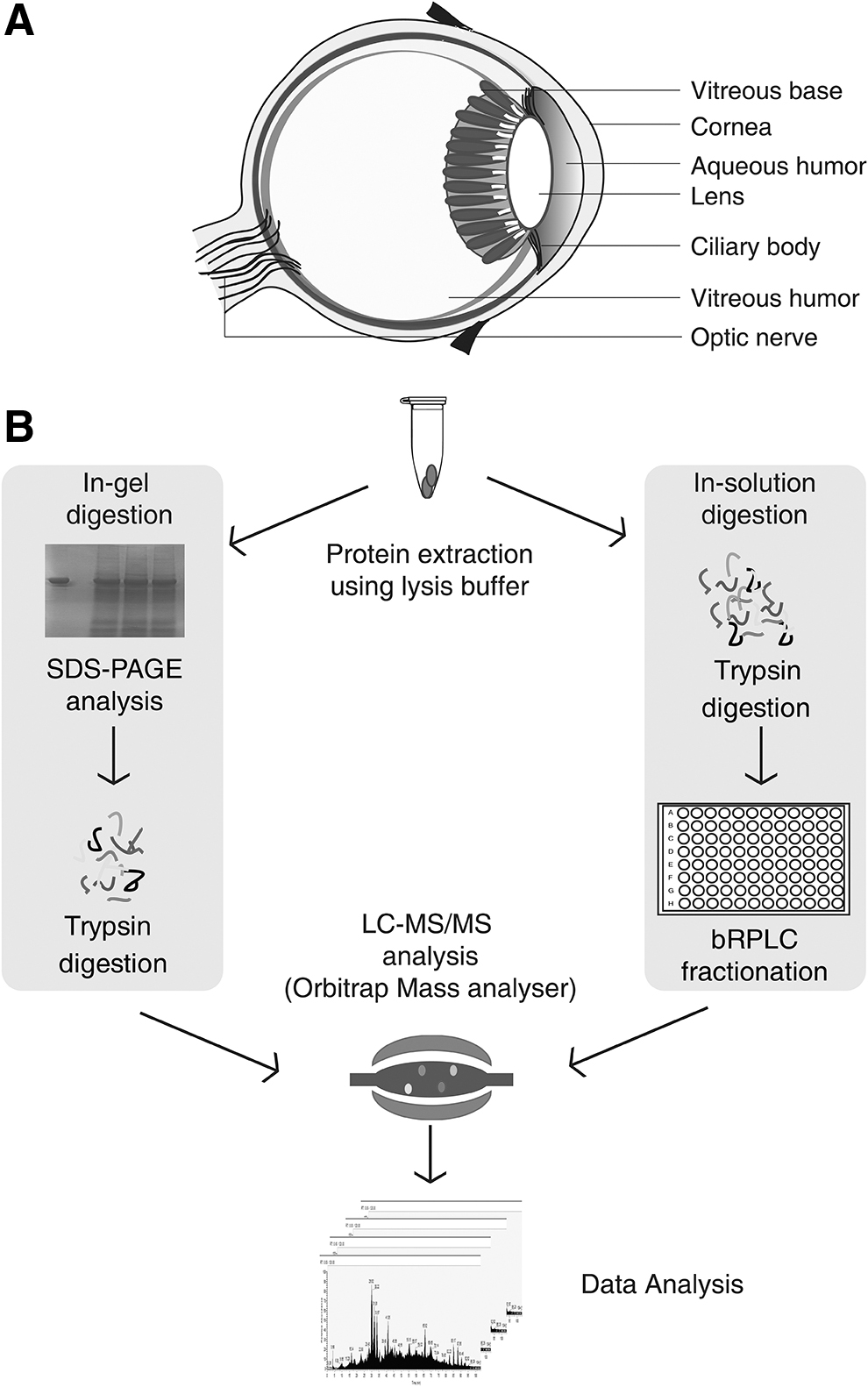
Schematic structure of the eye and an illustrated view of the experimental work carried out to profile the human vitreous base tissue
A Partial List of Proteins Identified from the Vitreous Base Along with Their Functions
To measure the relative abundance of the proteins in the vitreous base, we employed iBAQ analysis (Schwanhausser et al., 2011), which revealed albumin (ALB), crystallins (CRYBB2, CRYGS, CRYBA1, CRYAB, and CRYBB1), vimentin (VIM), histone proteins (HIST1H4H, HIST2H2BE, H3F3B, and HIST2H2AC), transferrin, and keratins (KRT1, KRT2, KRT9, KRT10, and KRT77) to be highly abundant of the total protein content. Interestingly, we identified several scaffold proteins also to be abundantly expressed. The entire list of proteins with their corresponding iBAQ values has been provided in Supplementary Table S4.
Gene ontology-based functional classification of proteins
To determine the various functions of the identified proteins, we carried out gene ontology-based classification using ProteinCentre Annotation node in Proteome Discoverer. The most enriched biological processes included metabolic (21.97%) and regulation of biological process (21.00%) (Fig. 2A). Similarly, the enriched molecular functions include protein binding (34%), catalytic activity (20%), and metal ion binding activity (10%) (Fig. 2B). Classification based on their subcellular localization revealed that the maximum number of proteins was localized to the membrane (21%), followed by cytoplasm (18%), nucleus (13%), and cytosol (13%) as depicted in Figure 2C.
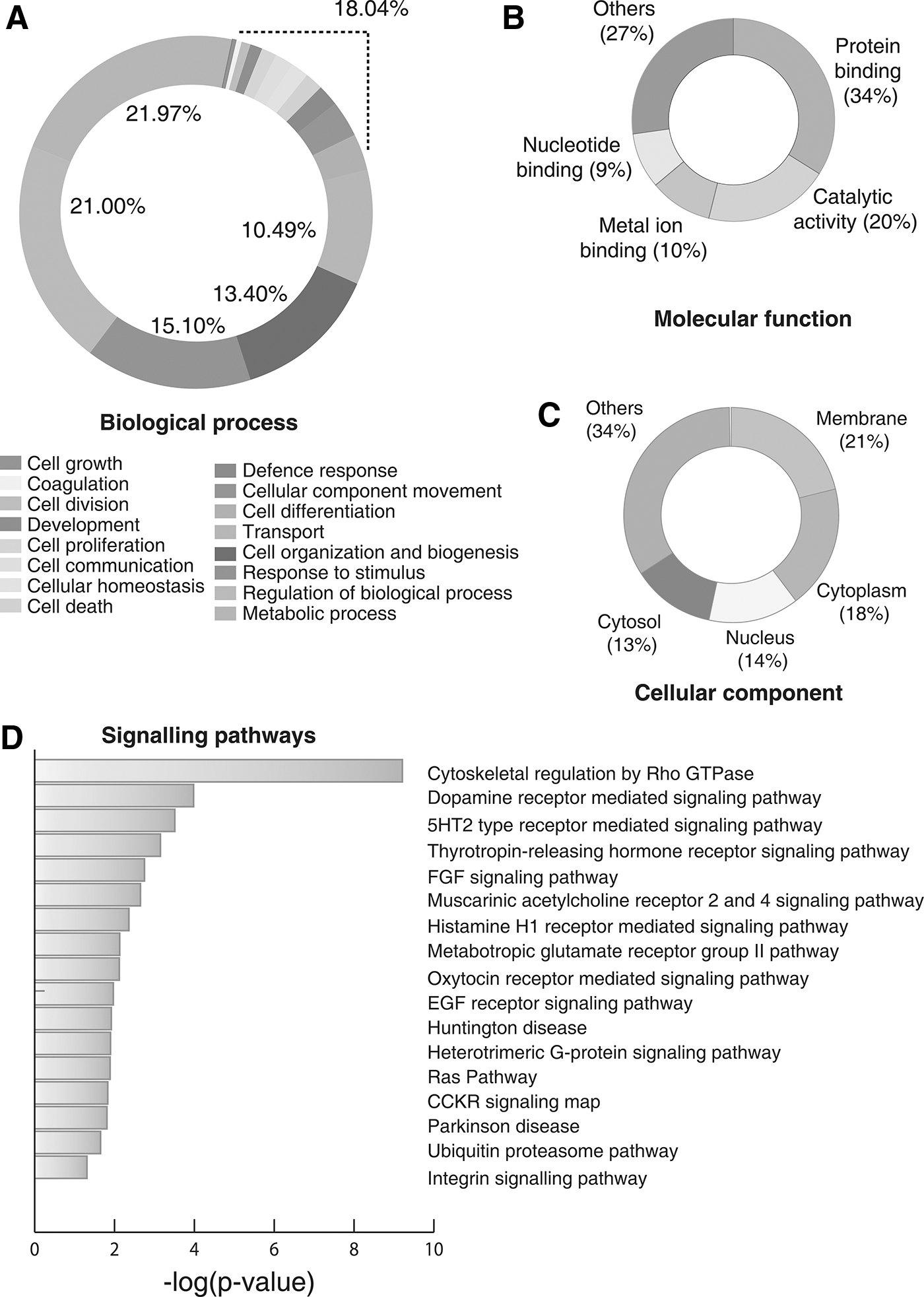
Gene Ontology analysis and Pathway analysis of the proteins identified in vitreous base,
Pathway analysis of the vitreous base proteome
Based on the cues, obtained from gene ontology analysis, we next performed pathway enrichment analysis using the PANTHER Overrepresentation Test (Annotation version PANTHER version 11.1 Released 2016-10-24). Our analysis revealed proteins involved in 15 pathways to be significantly enriched, including ubiquitin-proteasome pathway (UPP), histamine H1 receptor-mediated signaling pathway, metabotropic glutamate receptor group II pathway, integrin signaling pathway, Ras Pathway, and CCKR signaling map, among others. A list of statistically significant pathways enriched in this dataset is provided in Supplementary Table S5 and depicted in Figure 2D.
The UPP is a principal pathway that is involved in selective degradation of modified or aberrant proteins that are potentially cytotoxic in the eye and known to play a crucial role during eye development (Shang and Taylor, 2012). The process is mediated by ubiquitination, followed by proteasomal degradation. Therefore, this cytosolic proteolytic pathway is one of the vital cellular mechanisms that regulate various cellular processes, including cell signaling, cell cycle control, proliferation, development, and synaptic plasticity (Hegde, 2010).
Studies on the aberrant impairment in the UPP activity have demonstrated increased accumulation of abnormal proteins in the retina, implicating its role in the pathogenesis of age-related macular degeneration (Shang and Taylor, 2012). Histamine H1 receptor-mediated signaling pathway is mediated by one of the four histamine receptors, resulting in mobilization of calcium (Ca2+) (Li et al., 2012). These H1 receptors are known to be ubiquitous in their distribution and regulate cellular responses. Both H1 and H2 receptors are known to mediate vasodilator response, but the vascular permeability is mediated by H1 receptors only (Owen et al., 1980). In the eye, they are known to have a critical role in regulating sensory signaling (Van Ruitenbeek et al., 2009).
In addition, the identified proteins were enriched in cellular and metabolic pathways such as glycolysis, metabolic pathways, pyruvate metabolism, and focal adhesion. The proteins mediating these pathways play a critical role during the generation of reactive oxygen species (ROS), thus protecting the eye from oxidative stress as this is known to be associated with aging and inflammation (Hegde et al., 2010). As the tissues of the eye rely on glycolysis for its oxygen dependency and visual information processing, we identified proteins involved in such pathways (Hegde et al., 2010). Oxidative stress has been implicated in vitreoretinal disease, and the elevated levels of ROS are associated with different ocular maladies such as myopia, diabetic vitreoretinopathy, cataract, glaucoma, and uveitis (Bouhenni et al., 2011; Chen et al., 2009. Duan et al., 2008; Fujiwara, 1989; Fuchshofer and Tamm, 2009; Izzotti et al., 2010; Ladas et al., 2001; Satici et al., 2003; Sawada et al., 2009).
In this study, we identified proteins with known antioxidant activity such as superoxide dismutase 1 (SOD1), SOD2, SOD3, and arachidonate 5-lipoxygenase-activating protein (ALOX5AP), which play an essential role in preventing the eye from oxidative damage and any dysregulation of such proteins can result in oxidative stress-related vitreoretinopathies.
Advancement in proteomics technologies accelerated the kinome analysis. We attempted to decipher the signaling networks mediated by the human protein kinases as the aberration of kinase signaling has been found to be associated with a variety of diseases and drug resistance (Cohen, 2002; Zhang et al., 2009; Graves et al., 2013). Analysis of the kinome expression dynamics revealed the identification of 301 kinases in this study, where a vast majority was found to be associated with mitogen-activated protein kinase (MAPK) cascade. This pathway consists of serine/threonine kinases such as MAPK1, MAPK3, MAPK7, MAPK8, MAPK10, MAPK14, MAPKAPK2, and MAPKAPK3, which play a significant role in regulating the cellular process by enabling effector-receptor coupling (Supplementary Fig. S1).
Several studies report the activation of the MAPK cascade in trabecular meshwork, resulting in cellular apoptosis (Alexander and Acott, 2003; Hashimoto et al., 2005; Li et al., 2006). Similarly, protein tyrosine kinases (PTKs), identified in this study, such as tyrosine-protein kinase 7 isoform e (PTK7), proto-oncogene tyrosine-protein kinase (SRC), and tyrosine-protein kinase (CSK), are also known to be involved in ciliary muscle contraction (Wiederholt et al., 1998). Like kinases, phosphatases are known to be involved in oxidative stress regulation. A proper understanding of the role of kinases and phosphatases will aid in the identification of new targets for therapeutic interventions.
Proteins previously identified in other studies
It has been documented that the human vitreous is composed of several substructures, and each of them plays a vital role in the constitution of the ocular tissue, thereby suggesting distinct proteome profiles. Recently, Skie et al. (2015) carried out a proteomic analysis of four distinct anatomical regions of human vitreous, including vitreous core, vitreous base, vitreous cortex, and anterior hyaloid, and identified a mean of 2000 unique proteins in each of the substructure. Comparison of our findings with the substructure proteome revealed 4877, 4198, 5319, and 4818 proteins to be unique in comparison with vitreous base, cortex, core, and anterior hyaloid, respectively (Skeie et al., 2015).
PPI analysis was carried out using STRING (www.string-db.org) to identify the protein complexes playing a crucial role in biological processes. It was done using k-means clustering, which revealed a vast majority of proteins to be involved in the metabolic process and are cytoskeletal (Fig. 3A, B). We identified 796 proteins that have already been previously reported in the vitreous base (Skeie et al., 2015). This has been made possible by significant improvements in the technology, which has enabled an increase in both the peptide and protein identifications compared to the previous series of mass spectrometers. To the best of our knowledge, this study constitutes the most extensive catalog of the human vitreous base proteome identified to date.
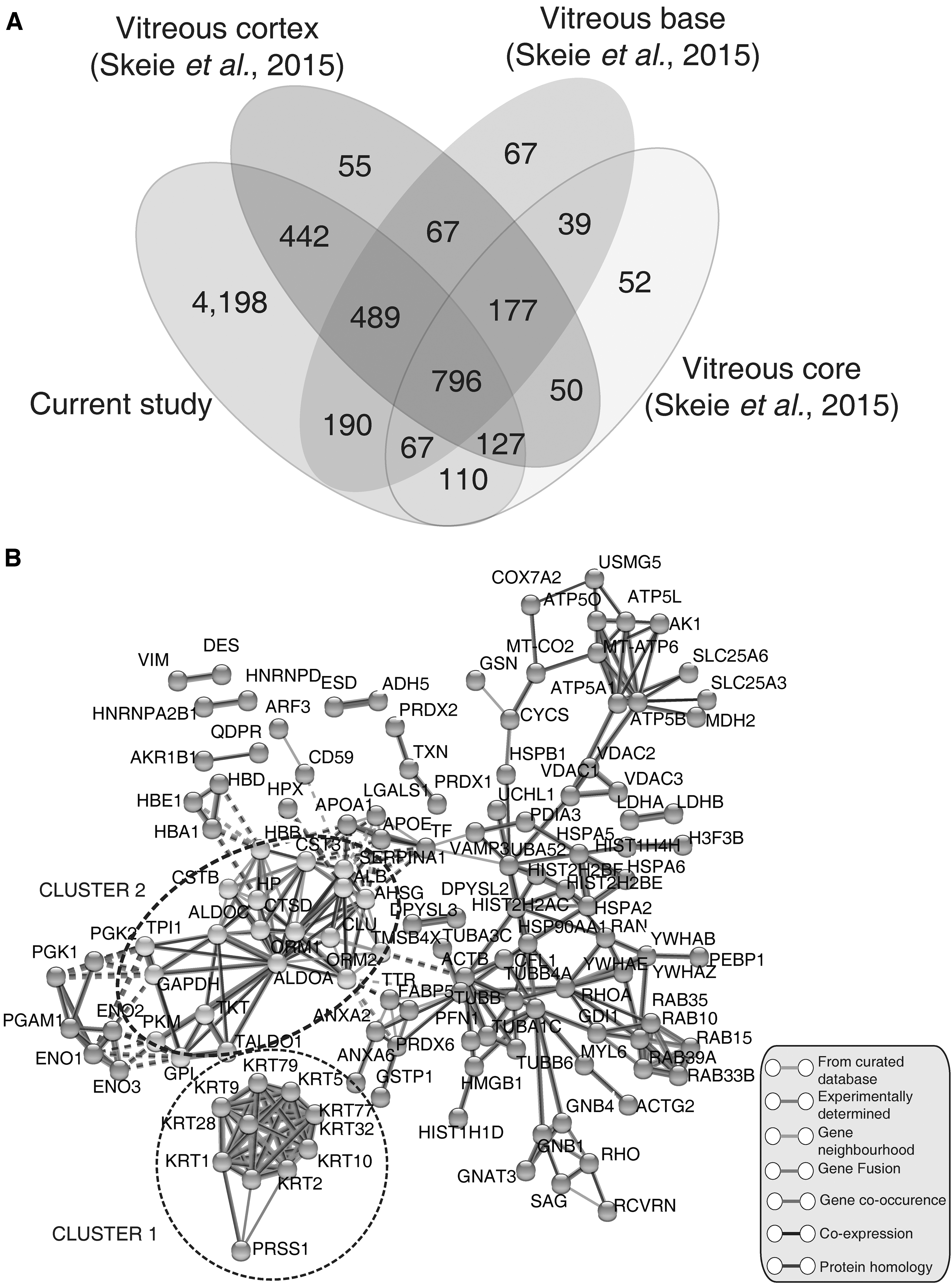
Comparison with previous datasets and PPI.
Furthermore, we also compared our data with the list of proteins classified as “missing proteins” by Chromosome-Centric Human Proteome Project (C-HPP) study (neXtProt release 2016-12-02) (Gaudet et al., 2017). Our analysis provides protein-coding evidence for three proteins RAS p21 protein activator 4B (RASA4B), RAB, member of RAS oncogene family-like 2B (RABL2B), and opsin 1, medium wave sensitive 3 (OPN1MW3) (Supplementary Fig. S2H–J). This analysis is based on the criteria for the classification of missing proteins (Paik et al., 2012).
Opsins are membrane proteins, found mostly in the photoreceptor cells such as rod cells and cone cells of the retina and are known to play a role in visual transduction (Chinen et al., 2003). OPN1MW3 is reported to be one of the cone opsins and known to encode green opsins (Shi et al., 2019). In 2005, a study reported the presence of the genes encoding this protein in zebrafish along with other visual opsins (Chinen et al., 2005).
In the same year, a study demonstrated the random arrangement of the cone cells encoding medium-wavelength opsins for vision in humans and their detrimental role in peripheral color vision (Hofer et al., 2005). RABL2B protein belongs to the RAS GTPase superfamily, where it is highly expressed in ciliated cells compared to nonciliated cells (Hoh et al., 2012). As cilated and flagellated cells exhibit a vital role in motility and sensory functions, any dysfunction in cilia impacts the normal phenotype and is known to cause retinal degeneration along with other abnormalities such as diabetes, obesity, and skeletal dysplasias (Kanie et al., 2017; Waters and Beales, 2011). The role of these proteins in the vitreous base remains to be determined.
Regulatory protein hubs in the vitreous base proteome
Proteins are known to exhibit wide array of structural and functional activities and play a detrimental role in maintenance of cellular architecture. These activities rarely allow proteins to act alone and therefore, to carry out such activities, they need to interact with other proteins or biomolecules. To understand such relationships of proteins, we carried out the network analysis of the of the 200 most abundant proteins to identify the hub proteins with regulatory roles where the protein-protein network was generated using STRING. This information was then overlayed in Cytoscape, where the topology parameters were generated using NetworkAnalyzer, and their functions were explored (Fig. 4A, B, Supplementary Tables S6 and S7).
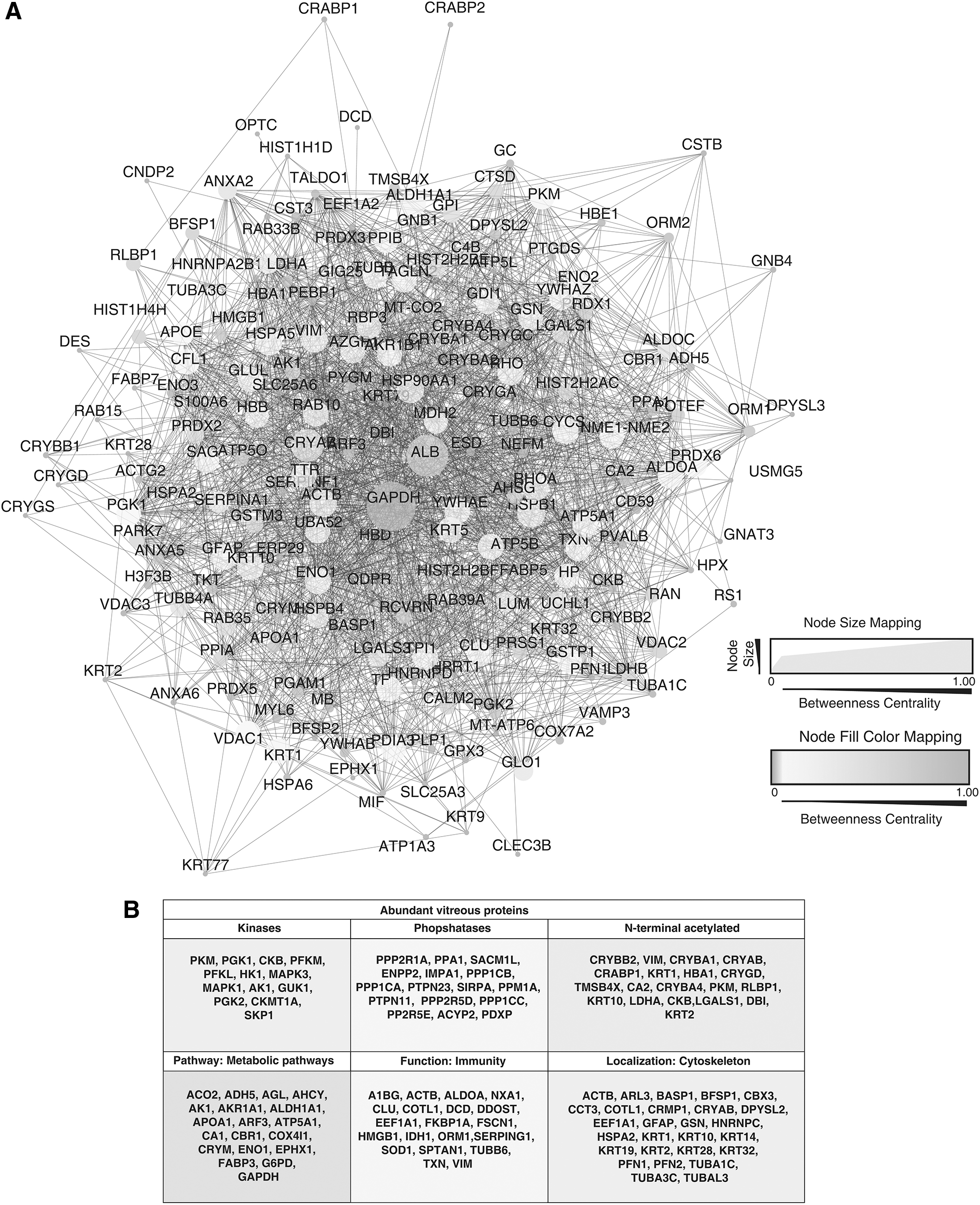
Network analysis and functional importance of the top abundant proteins identified in vitreous base.
The betweenness centrality resulted in the identification of the hub proteins, which were represented by high betweenness centrality scores. The proteins such as GAPDH, ALB, CRYAB, HSPB1, ACTB, HSP90AA1, KRT10, RHOA, VIM, ENO1, RHO, CYCS, and HSPA5 were represented with high scores. Heat shock proteins are known to protect the cellular proteins and maintain cellular viability under stressful situations (Hartl and Hayer-Hartl, 2002; Han et al., 2008). This analysis identified heat shock proteins such as HSPB1, HSP90AA1, and HSPA5 in the vitreous base. Reports demonstrate that the aberrant expression of such proteins during ocular stress can serve as a cytoprotective response (Georgopoulos and Welch, 1993).
Similarly, we also identified Crystallin Alpha B (CRYAB) as one of the important regulatory proteins. It is a well-known molecular chaperone that plays a significant role in maintaining the intracellular architecture by binding misfolded proteins and preventing aggregation (Arrigo et al., 2007). It is mainly found in the lens where it acts as a refractive element and provides ocular stability (Urbak and Vorum, 2010).
Conclusions
Proteomic investigation using high-resolution mass spectrometry serves as a powerful method for profiling proteins involved in many pathophysiological processes. This study provides a proteomic landscape of the human vitreous base using a high-resolution Orbitrap platform in an unbiased manner. We report here an additional 4198 proteins with respect to the previous studies. Several ocular pathologies implicate the involvement of the vitreous; however, there is insufficient information about the role of such molecules involved in disease pathophysiology.
Further investigation involving healthy and disease states would be necessary to identify such actionable targets and unravel their pathogenic mechanism. The proteomic insights will aid in achieving a better understanding at the molecular level, and further research can be carried to unravel its role in diseased states. These data also invite us to consider a potentially more active functional role for the vitreous base in eye physiology and visual health.
Footnotes
Acknowledgments
We thank Karnataka Biotechnology and Information Technology Services (KBITS), Government of Karnataka, for the support to the Center for Systems Biology and Molecular Medicine at Yenepoya (Deemed to be University) under the Biotechnology Skill Enhancement Programme in Multiomics Technology (BiSEP GO ITD 02 MDA 2017). We thank Yenepoya (Deemed to be University) for access to instrumentation. Varshasnata Mohanty is a recipient of the Women Scientist-A award from the Department of Science and Technology (DST), Government of India. Sneha M. Pinto is a recipient of INSPIRE Faculty Award from DST, Government of India. Mohd Altaf Najar is a recipient of the Senior Research Fellowship from University Grants Commission (UGC), Government of India.
Authors' Contributions
S.M.P., K.R.M., and T.S.K.P. conceptualized the study, designed the experiments, and reviewed the article. K.B.M. and K.R.M. provided vitreous base samples for the study. S.M.P., M.A.N., and V.M. carried out sample preparation, fractionation, and mass spectrometry. V.M., S.M.P., and Y.S., analyzed the mass spectrometry data. V.M., S.M.P., Y.S., K.B.M., K.R.M., and T.S.K.P. wrote the article. All the authors read and revised the article for important intellectual content, and approved the final article.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
The authors received no financial support for the research, authorship, and/or publication of this article.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.