
Abstract
The tumor stroma, a key component of the tumor microenvironment (TME), is a key determinant of response and resistance to cancer treatment. The stromal cells, extracellular matrix (ECM), and blood vessels influence cancer cell response to therapy and play key roles in tumor relapse and therapeutic outcomes. Of the stromal cells present in the TME, much attention has been given to cancer-associated fibroblasts (CAFs) as they are the most abundant and important in cancer initiation, progression, and therapy resistance. Besides releasing several factors, CAFs also synthesize the ECM, a key component of the tumor stroma. In this expert review, we examine the role of CAFs in the regulation of tumor cell behavior and reveal how CAF-derived factors and signaling influence tumor cell heterogeneity and development of novel strategies to combat cancer. Importantly, CAFs display both phenotypic and functional heterogeneity, with significant ramifications on CAF-directed therapies. Principal anti-cancer therapies targeting CAFs take the form of: (1) CAFs' ablation through use of immunotherapies, (2) re-education of CAFs to normalize the cells, (3) cellular therapies involving CAFs delivering drugs such as oncolytic adenoviruses, and (4) stromal depletion via targeting the ECM and its related signaling. The CAFs' heterogeneity could be a result of different cellular origins and the cancer-specific tumor microenvironmental effects, underscoring the need for further multiomics and biochemical studies on CAFs and the subsets. Lastly, we present recent advances in therapeutic targeting of CAFs and the success of such endeavors or their lack thereof. We recommend that to advance global public health and personalized medicine, treatments in the oncology clinic should be combinatorial in nature, strategically targeting both cancer cells and stromal cells, and their interactions.
Introduction
Recent cancer incidence and mortality statistics indicate an always increasing burden of cancer in the coming years, with most new cases occurring in countries of low and middle income (Bray et al., 2018; Ferlay et al., 2019; Fidler and Bray, 2018). Based on GLOBOCAN estimates and others, there is a need for a new drive to find and develop new strategies to reduce this burden worldwide ( Bray, 2016; Bray and Soerjomataram, 2015; Bray et al., 2012, 2018; Ferlay et al., 2019; Fidler et al., 2016). The cancer incidence and mortality statistics provided are likely lower than the actual figures given the limited and strained surveillance systems found in many low- and medium-income countries (Bray et al., 2018; Ferlay et al., 2019; Znaor et al., 2018). It is important to note that progress has been made in raising awareness to cancer and several cancer prevention strategies are being adopted; however, the picture still looks gloomy going forward.
Although great success has been achieved in treating cancer via different therapeutic strategies directed at cancer cells, reports show that cancer deaths are likely to increase globally (Bray and Soerjomataram, 2015; Bray et al., 2018; Ferlay et al., 2019; Fidler et al., 2016; Moten et al., 2014). Significant progress made in understanding the underlying causes and molecular mechanisms involved in tumor initiation and development has led to better patients' outcomes (Lesina et al., 2011; Mongiat et al., 2016; Senthebane et al., 2017, 2018). Two major contributors to cancer deaths still requiring better understanding are cancer relapse and metastasis (Crunkhorn, 2018; Li et al., 2015; Yates et al., 2017). Research into tumor relapse and metastasis is ongoing, with several studies reporting novel drugs and effective strategies aimed at limiting these processes (Mitra et al., 2015; Zhou et al., 2009).
Further, studies have revealed the tumor microenvironment (TME) as key to tumor initiation, response to therapy, relapse, and metastasis, ultimately influencing patients' management in the clinic (Dzobo et al., 2016a; Kalluri, 2016; Senthebane et al., 2017, 2018). Specifically, the TME has been shown to provide some form of protection to cancer cells, reduce cancer cell response to therapy, and, ultimately, promote therapy resistance (Quail and Joyce, 2013; Senthebane et al., 2017, 2018). The TME can also modify cancer cells, resulting in cancer cell heterogeneity (Dzobo et al., 2018b; Quail and Joyce, 2013).
Studies have shown that subpopulations of cells within the TME, including cancer-associated fibroblasts (CAFs), can affect cancer cells differently (Ishii et al., 2016; Öhlund et al., 2014, 2017). In addition, several studies have shown that the extracellular matrix (ECM) can influence cancer cell proliferation, migration, and response to therapy (Noguera et al., 2012; Senthebane et al., 2018). This has resulted in increased attention being given to the role of tumor-associated cells and ECM as well as the development of therapies directed at the tumor stroma. Recently, studies also demonstrated the gut microbiota's influence on cancer response to therapy through modulation of TME (Iida et al., 2013; Vétizou et al., 2015).
While underscoring the importance of microbiota in disease treatment and outcome, these studies show that modulation of TME play a huge part in cancer cell response to treatment. In addition, the activation of the immune system has been shown to influence cancer cell sensitivity to radiotherapy (Strom et al., 2017). Thus, the targeting or modulation of the TME has real therapeutic value with the potential to improve patients' outcomes. A deeper understanding of the TME and its contribution to cancer cell behavior is pertinent. This comprehensive review presents advances in our knowledge on CAFs and the role played by these cells in disease initiation and progression. This review brings to the fore the strategies being adopted to include TME-directed therapies in the clinic, with the aim of reducing fatal disease.
The TME is used to describe the cells, ECM, and blood vessels found within the vicinity of cancer cells in a tumor. Included in this definition are cells such as fibroblasts, macrophages, endothelial cells, lymphocytes, myeloid-derived cells, and the ECM. Several studies have shown that depending on the stage of tumor development, the TME can provide both an inhibitory and promoting environment to cancer cell growth (Senthebane et al., 2017, 2018). Cells within the TME can be from the local vicinity or migrated from distant environments (Friedl and Alexander, 2011; Wels et al., 2008).
The CAFs are the most abundant cells within the TME and play key roles in tumor initiation and progression (Kalluri, 2016; Su et al., 2018). Immune cells within the TME are mostly macrophages (Hao et al., 2012; Kim et al., 2012). Endothelial cells mostly function to form new blood vessels that are necessary for the supply of nutrients to the growing tumor and removal of toxic waste (De Palma et al., 2017). In addition, endothelial cells also secrete platelet-derived growth factor (PDGF), which attracts pericytes to tumor blood vessels, and these pericytes are involved in the stabilization of newly formed blood vessels (Armulik et al., 2011).
Stromal cells such as fibroblasts and macrophages are also involved in the synthesis and maintenance of the ECM (Bonnans et al., 2014; Riches, 1988). By releasing enzymes such as matrix metalloproteinases, and growth factors, stromal cells contribute to tumor ECM remodeling (Bonnans et al., 2014). ECM remodeling enables cancer cells to migrate and invade surrounding tissues (Gaggioli et al., 2007). As the tumor develops, both tumor cells and the TME components co-evolve and are transformed through the release of various growth factors and other biomolecules, with the TME initially being tumor-restrictive but tumor-promoting at later stages (Senthebane et al., 2017, 2018).
Several studies have demonstrated stromal cell heterogeneity, especially of CAFs and macrophages (Bauer et al., 2010; Öhlund et al., 2017; Su et al., 2018). This heterogeneity manifests as either tumor-restrictive or tumor-promoting activities of the cells (Özdemir et al., 2014; Rhim et al., 2014). Elaborate studies by Su et al. as well as by Öhlund et al. demonstrated the presence of distinct populations of fibroblasts within tumors that have specific functions and that they influence cancer cell response to therapy (Hwang et al., 2008; Öhlund et al., 2017; Su et al., 2018).
Lately, the use of cell surface markers to isolate and characterize these distinct population of fibroblasts has allowed a deeper analysis of their behavior and functions (Öhlund et al., 2017; Su et al., 2018). The identification and use of specific cell surface markers can allow targeted manipulation of specific fibroblasts populations to achieve a specific goal during cancer treatment. Under normal physiological conditions, fibroblasts are dormant/quiescent or inactive but can be activated by various growth factors, cytokines, and chemokines (Gaggioli et al., 2007; Mueller and Fusenig, 2004).
Currently, very few clinical trials have been conducted that target CAFs to treat cancer. This is partly because the translation of laboratory scientific evidence into clinical use requires more resources. It is, therefore, imperative that a deeper understanding of stromal cell behavior and biology is obtained to improve strategies targeting such cells. This review describes in detail the involvement of CAFs in cancer pathogenesis.
Mapping CAFs' Origins and Heterogeneity
In a landmark publication in 1858, Virchow described fibroblasts as cells that were spindle shaped and were responsible for the synthesis of collagen (Virchow, 1871). Further studies showed that fibroblasts are mostly dormant cells within the ECM and are transformed under conditions such as inflammation, fibrosis, and wound healing (Darby and Hewitson, 2007; Kalluri and Zeisberg, 2006). With both inflammation and fibrosis being associated with cancer initiation and development, fibroblasts are, therefore, activated during these processes (Dvorak, 1986; Hanahan and Weinberg, 2011; Wynn and Ramalingam, 2012). These activated fibroblasts associated with cancer are referred to as CAFs or tumor-associated fibroblasts (Kalluri and Zeisberg, 2006; Mueller and Fusenig, 2004).
Although initially having an anti-tumorigenic phenotype as normal fibroblasts, CAFs eventually become pro-tumorigenic through mechanisms that are still under intense investigations (Öhlund et al., 2014; Senthebane et al., 2017). The CAFs will eventually become the dominant stromal cell type within the TME and promote tumor progression via release of several factors and the synthesis of the ECM (Hanahan and Weinberg, 2011; Kalluri, 2016; Senthebane et al., 2017, 2018).
Irrespective of their cell of origin, CAFs are large spindle-shaped cells showing increased stress fibers and well-developed cellular-ECM connections (De Wever et al., 2008). Although their shape appears identical to normal fibroblasts, CAFs show increased numbers of ribosomes and a rough endoplasmic reticulum (De Wever et al., 2008). Early markers used for positive identification of CAFs include alpha-smooth muscle actin (α-SMA), vimentin, and desmin (Lazard et al., 1993; Znaor et al., 2018). Unlike their normal equivalent, CAFs demonstrate increased proliferation and migration capacity and show increased expression of ECM proteins and ECM degrading enzymes (Ma et al., 2009; Saadi et al., 2010). The resulting remodeling of the TME, referred to as desmoplasia, causes fibrosis and stiffening of the tissue (Kalli and Stylianopoulos, 2018; Poltavets et al., 2018).
The presence of CAFs and tissue stiffening have been associated with cancer relapse, implying that CAFs and desmoplasia contribute to cancer progression and therapy resistance (Laklai et al., 2016; Tsujino et al., 2007). Calvo et al. (2013) demonstrated that ECM remodeling is a requirement for CAFs' continuous presence within the TME. Thus, there is a feedback loop whereby CAFs build and maintain the TME and remodeling of the ECM whereas the remodeling is needed for both the generation and maintenance of CAFs (Calvo et al., 2013; Maller et al., 2013).
The balance between ECM synthesis and degradation is needed for homeostasis maintenance. ECM proteins such as collagens and fibronectin can block immune cells from infiltrating into the tumor (Cukierman and Bassi, 2010). In addition, the ECM provides the “theater” in which all the cellular interactions take place; blood vessels are formed; as well as cancer cells are allowed to escape immune detection (Cukierman and Bassi, 2010; Gorski et al., 1994).
The CAFs promote angiogenesis through the release of MMPs, which degrade the ECM and allow the release of vascular endothelial growth factor-A (VEGF-A) sequestered by the ECM (Baeriswyl and Christofori, 2009). The release of VEGF-A promotes the formation of the vascular system, allowing the tumor to grow large with enhanced exchange of nutrients and toxic substances (Baeriswyl and Christofori, 2009; Raica et al., 2009). The CAFs are known to release several growth factors and cytokines that are known to promote inflammation and to assist in the evasion of the immune system (Flavell et al., 2010; Yang et al., 2016). In short, CAFs are the builders and are involved in the maintenance of the TME via synthesis and release of ECM proteins and protein factors (De Wever et al., 2008; Hwang et al., 2008; Kalluri, 2016; Kalluri and Zeisberg, 2006; Senthebane et al., 2017, 2018).
In turn, the TME promotes tumor initiation, progression, and metastasis (De Wever et al., 2008; Hanahan and Weinberg, 2011; Kalluri, 2016; Kalluri and Zeisberg, 2006; Öhlund et al., 2014). Increased knowledge on the origins and role of CAFs within the TME may allow the development of new anti-cancer strategies (Brechbuhl et al., 2017; Bussard et al., 2016; Casey et al., 2008; Glentis et al., 2017).
Several studies show that CAFs originate from different cell types and this has been suggested to cause the heterogeneity observed in these cells (De Wever et al., 2008; Kalluri and Zeisberg, 2006; Öhlund et al., 2014; Senthebane et al., 2017). Besides the activation of fibroblasts within the vicinity of the cancer cells, CAFs can also originate from bone marrow-derived fibrocytes and mesenchymal stem/stromal cells (MSCs) that are recruited to the tumor; the transdifferentiation of pericytes, smooth muscle cells, and adipocytes; epithelial cells that underwent epithelial to mesenchymal transition (EMT); endothelial cells that underwent endothelial to mesenchymal transition (EndMT); and, finally, the activation of quiescent stellate cells (Fig. 1) (Öhlund et al., 2017; Omary et al., 2007; Yin et al., 2013).
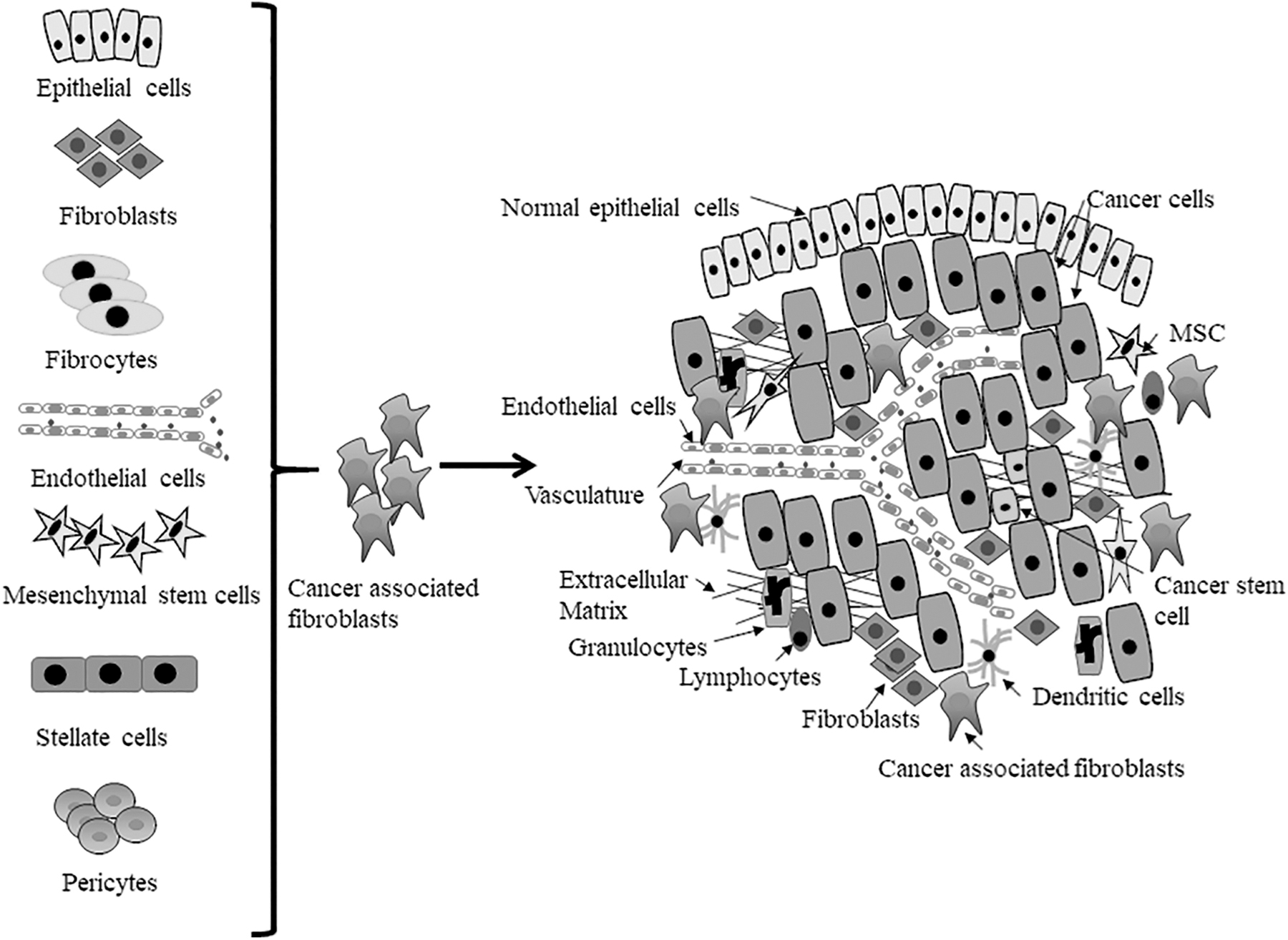
CAFs can be derived from different cellular sources. Fibroblasts and stellate cells can be activated through various processes to become CAFs; mesenchymal stem cells and fibrocytes are recruited to the tumor via circulation; epithelial cells and endothelial cells undergo epithelial to mesenchymal transition and endothelial to mesenchymal transition, respectively, to become CAFs. Further, pericytes and adipocytes are known to undergo transdifferentiation to become CAFs. CAFs, cancer-associated fibroblasts; MSC, mesenchymal stem cell. Figure is adapted from our earlier publication, Senthebane et al. (2017).
As demonstrated in breast cancer, bone marrow-derived fibrocytes that are usually inactive and present in circulation can be recruited to the tumor and become CAFs over time (Barth et al., 2002).
Our recent work showed that MSCs can be converted to “CAFs” over time through interaction with cancer cells (Senthebane et al., 2017). We demonstrated that transformation of MSCs into CAFs was dependent on the release of transforming growth factor-beta (TGF-β) by both cancer cells and the MSCs (Senthebane et al., 2017). Several other studies have also shown that MSCs can transform/differentiate into CAFs in several cancers (Jung et al., 2013; Zhu et al., 2014). For example, Weber et al. (2015) demonstrated that osteopontin mediates TGF-β-dependent transformation of MSCs into CAFs in breast cancer.
Tissue-resident MSCs and those recruited from distant tissues and organs can interact with cancer cells and be transformed to CAFs. Quiescent stellate cells can also transform into CAFs when activated and contribute to the CAF population in the liver and the pancreas (Omary et al., 2007; Yin et al., 2013). Epithelial cells within the vicinity of cancer cells can undergo EMT and become CAFs under fibrotic conditions (Iwano et al., 2002). This means that epithelial-derived cancers can end up with a huge population of CAFs driving tumor progression. Endothelial cells can also undergo EndMT and become CAFs (Zeisberg et al., 2007). Both transformed epithelial cells and endothelial cells express CAF markers, including S100A4 (Iwano et al., 2002; Zeisberg et al., 2007).
Through a transdifferentiation process, cells such as adipocytes and pericytes can become CAFs (Dulauroy et al., 2012; Jotzu et al., 2011). It is, therefore, plausible to speculate that all cells that are within the vicinity of cancer cells and those that end up at the tumor site via circulation can potentially be transformed into cells that will eventually promote tumor growth such as CAFs. We are just beginning to get a clear picture of the components of the TME and functions in tumor initiation and progression, and detailed mechanistic studies are likely to reveal interesting information.
It is now accepted that CAFs show heterogeneity with distinct subsets of cells displaying different phenotypes and functions within the TME (Öhlund et al., 2014, 2017; Orimo and Weinberg, 2007; Su et al., 2018). Further, CAFs' heterogeneity is also dependent on the stage of tumor development (Huelsken and Hanahan, 2018). Normal fibroblasts are generally considered genetically stable, whereas the same cannot be said about CAFs. The process of transformation, the generation of reactive oxygen species (ROS), and the hypoxic conditions within the tumor can result in DNA alterations in CAFs as well, with CAFs co-evolving with cancer cells as the tumor develops (Campbell et al., 2011; Öhlund et al., 2014).
Although rare, alterations to the genetic material of CAFs or stromal cells have been found in different cancers (Moinfar et al., 2000; Patocs et al., 2007). Several available techniques, including the use of CRISPR-Cas9 technology and tracing experiments, can be used to determine whether genetic alterations occur in CAFs as they do in cancer.
It is plausible to speculate that as the tumor evolves so do the cells associated with it, such as CAFs. Our study clearly demonstrates that previously anti-cancer activity of MSCs is diminished over time and “transformed” cells eventually become pro-tumorigenic (Senthebane et al., 2017). In addition, the synthesis of the ECM over time by both CAFs and cancer cells results in combinatorial ECM, which is pro-tumorigenic than normal ECM (Senthebane et al., 2018). Clearly, more research and focused analysis of the TME components is needed, especially the tumor-associated cells.
Given their possible cellular origin, it is not surprising that CAFs display great phenotypic heterogeneity. Different CAFs' subsets have been identified within the TME and are spatially distributed throughout the tumor (Öhlund et al., 2014; Patocs et al., 2007). Recent characterization of CAFs has identified specific biological markers for specific subsets of CAFs (Kalluri, 2016; Vennin et al., 2019). In addition, no CAF marker is expressed by all CAFs. Early studies identified markers such as α-SMA, fibroblast activation protein-alpha (FAP-α or just FAP), and PDGF receptor-β (PDGFR-β) to be expressed by different subsets of CAFs, with none being expressed by all CAFs (Akrish et al., 2016; Kalluri, 2016). To be useful, a combination of these markers is usually used during CAFs' characterization.
Among the markers, α-SMA has proven useful in the identification of CAFs as well as a marker for smooth muscle cells and pericytes (Öhlund et al., 2014). Senthebane et al. (2017) used both vimentin and α-SMA as markers to study cancer cell-derived TGF-β-mediated transformation of MSCs to CAFs in vitro. Over time, the expression of both markers was significantly increased in MSCs co-cultured with cancer cells. Cancers in which CAFs express elevated levels of α-SMA include breast, pancreatic, and liver cancers (Ayala et al., 2003; Yin et al., 2013). However, several other cells, including normal fibroblasts, smooth muscle cells, cardiomyocytes, and pericytes, also express α-SMA. Mostly involved in the maintenance of cellular cell structure, α-SMA is also involved in the migration and contraction of cells.
Another CAF marker vimentin is highly expressed by CAFs in breast and prostate cancers (Kalluri and Zeisberg, 2006; Vuoriluoto et al., 2011). We recently showed that vimentin can be induced via TGF-β-mediated transformation of MSCs (Senthebane et al., 2017). Biological functions of vimentin include promotion of migration and maintenance of cellular structure and integrity (Guo et al., 2013; Toivola et al., 2005; Wang and Stamenovic, 2002). In contrast to α-SMA and vimentin, desmin is mostly downregulated in CAFs and is also expressed by fibroblasts, muscle cells, and pericytes (Armulik et al., 2011; Eyden, 2008).
Caveolin-1 is a scaffolding protein expressed in CAF subpopulations as well as in cells such as endothelial cells, fibroblasts, and adipocytes. Low expression of caveolin-1 is used as a marker of a CAF subpopulation undergoing metabolic reprogramming and promoting tumorigenesis (Guido et al., 2012). In contrast, elevated levels of caveolin-1 are observed in CAFs with the propensity to promote metastasis (Cohen et al., 2004; Goetz et al., 2011). This clearly demonstrates the challenges involved in sorting and isolating of CAFs, a necessary step to their characterization, with huge ramifications to their therapeutic targeting.
Recently, CD10 and G-protein-coupled receptor 77 (GPR77) have been shown to be highly expressed in CAFs and are involved in promoting cancer stemness and resistance to chemotherapy in breast cancer cells (Su et al., 2018). Illustrating the challenges faced by any CAFs-targeted therapies, CD10 and GPR77 are also expressed in bone marrow-derived stromal cells and polymorphonuclear neutrophils, respectively (Karnoub et al., 2007; Su et al., 2018).
S100A4, sometimes referred to as fibroblast-specific protein, is expressed by fibroblasts within tumors and displays serine protease activity, allowing it to remodel the ECM (Zhang et al., 2013a). Fibroblasts expressing S100A4 are known to protect cancer cells via ECM production. Cells expressing S100A4 are known to be highly malignant and display a propensity for migration (Wang et al., 2005); for example, S100A4 is highly expressed in CAFs in breast cancers (Orimo et al., 2005).
This marker is also expressed by other cells, including cells undergoing EMT, macrophages, and normal fibroblasts (Li et al., 2010; Orimo et al., 2005; Zhang et al., 2013a). PDGFR-β is a CAF marker that has been targeted with kinase inhibitors (Pietras et al., 2008). PDGF receptor signaling inhibition with Imatinib was shown to abrogate malignant progression of cervical lesions (Pietras et al., 2008). CAFs found in colorectal and cervical cancers express high levels of PDGFR-β (Peña et al., 2013; Pietras et al., 2008). Macrophages have been shown to cause immune suppression through the expression of FAP (Arnold et al., 2014). The FAP is expressed in many human cancers and is highly expressed in CAFs (Arnold et al., 2014; Huber et al., 2003).
In summary, CAFs are a heterogeneous population of cells clearly demonstrating their diverse cellular origin and have been shown to have many functions within the solid tumor. A major challenge facing scientists today is the identification of reliable cell surface markers that can be used for therapeutic targeting. New data demonstrating that CAFs' function is dependent on the location within the tumor amplify the challenge faced (Öhlund et al., 2014, 2017). Irrespective of cell of origin, tumor stage, and location within the tumor, recent studies emphasize the need to identify cancer-specific CAF markers or CAF subset markers for use in diagnosis and anti-cancer targeting.
CAFs and Carcinogenesis
Data from several studies illustrate the supporting role of TME components in tumor progression, metastasis, and formation of new tumors (Bonnans et al., 2014; Cukierman and Bassi, 2010; Senthebane et al., 2017). Although initially being prohibitive of tumor growth, we demonstrated that over time cells within the TME promote tumor growth via the release of growth factors and deposition of ECM (Nicosia et al., 1993; Senthebane et al., 2017, 2018).
Several other studies confirmed that normal fibroblasts show inhibitory effects on cancer cell growth in vitro (Stoker et al., 1966). The expression of phosphatase and tensin homolog in stromal cells was shown to be necessary for this inhibition effect on epithelial tumors (Trimboli et al., 2009). Several growth factors and cytokines, including vascular endothelial growth factor (VEGF), stromal derived factor 1 (SDF-1), TGF-β, and interleukin-6 (IL-6), have been shown to be necessary in the transformation of normal stromal cells into cancer-supporting cells (Giannoni et al., 2010; Senthebane et al., 2017).
In addition, conditions within the TME such as hypoxia and ROS within the TME milieu also contribute to the conversion of normal cells to pro-tumorigenic cells (Maxwell et al., 1997; Ryan et al., 2000). Through the expression and secretion of growth factors and cytokines and their receptors, CAFs impact cancer cell-associated processes such as inflammation, angiogenesis, migration, invasion, chemoresistance, and immune evasion (Fig. 2) (Casey et al., 2008; San Francisco et al., 2004; Wang et al., 2017). Erez et al. (2010) demonstrated that normal fibroblasts can be induced to express inflammatory genes by carcinoma cells.
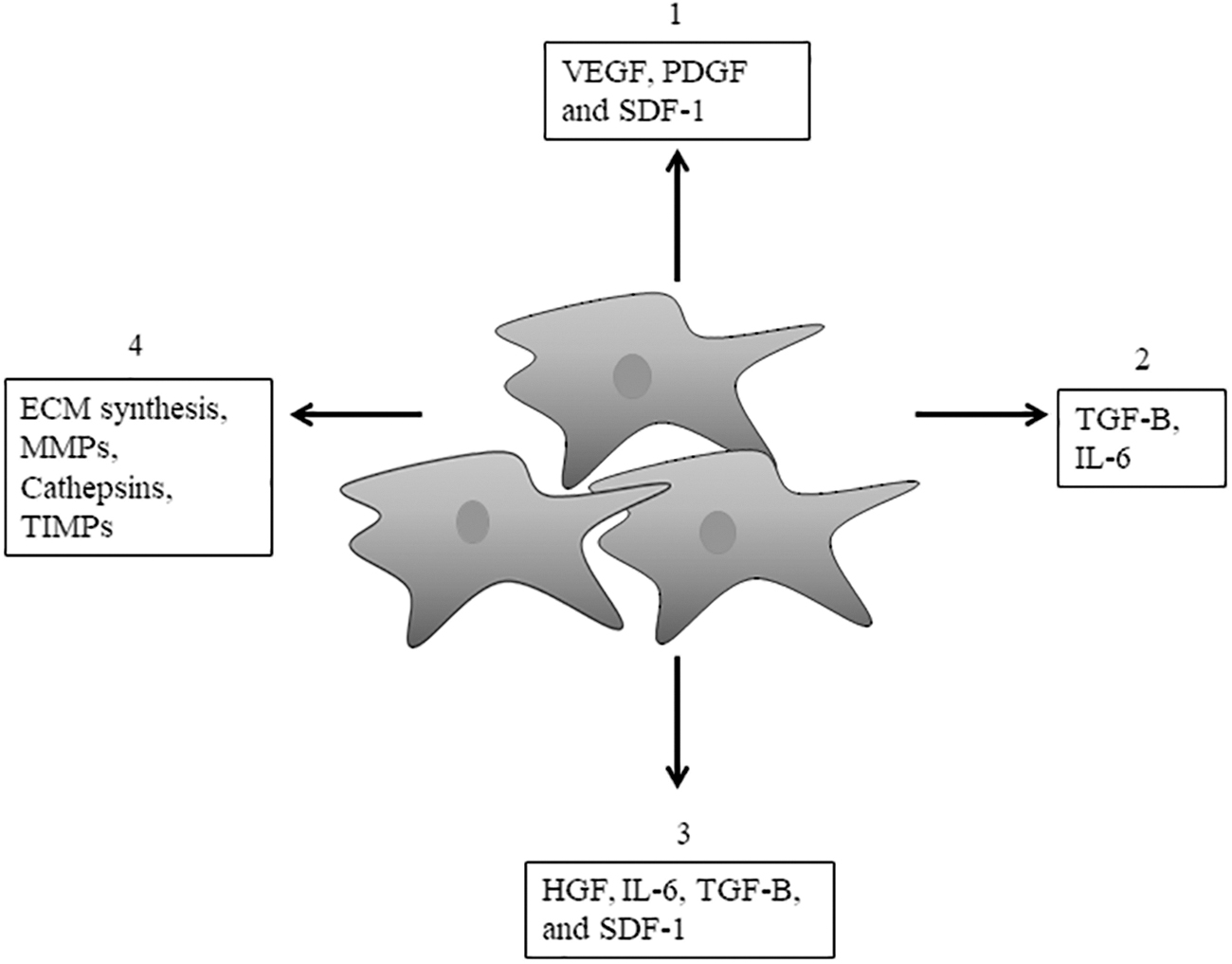
Effect of CAFs on tumor cells, other stromal cells, and the ECM within the TM. The CAFs synthesize and secrete the ECM, MMPs, and various factors, impacting cancer cell processes. Most of the secreted factors are growth factors and cytokines, including HGF, FGF, TGF-β, IL-6, IL-1, COX-2, VEGF, HIF1-α, PDGF, and SDF-1, and they impact tumor processes such as angiogenesis (1), chemoresistance (2), proliferation (3), and ECM remodeling (4). ECM, extracellular matrix; FGF, fibroblast growth factor; HGF, hepatocyte growth factor; HIF1-α, hypoxia inducible factor 1-α; IL-6, interleukin-6; MMPs, matrix metalloproteases; PDGF, platelet-derived growth factor; SDF-1, stromal-derived factor 1; TGF-β, transforming growth factor-beta; TIMP, tissue inhibitor of metalloproteinases; TME, tumor microenvironment; VEGF, vascular endothelial growth factor.
microRNAs (miRNAs) are also believed to be involved in the transformation of normal fibroblasts into CAFs. miRNAs are delivered to their target cells via exosomes, allowing miRNAs to induce and transform local and distant fibroblasts (Kahlert and Kalluri, 2013; Webber et al., 2010). A recent study by Fang et al. (2018) demonstrated that exosomal delivered miRNA-1247 from cancer cells induces the transformation of fibroblasts within lung premetastatic environments, thereby promoting metastasis.
Mitra et al. (2012) also demonstrated that cancer cells reprogram fibroblasts to CAFs via the release of miRNAs in ovarian cancer. Pang et al. (2015) also showed that cancer cell-derived miRNA-155 was involved in the transformation of normal fibroblasts into CAFs in pancreatic cancer. It is important to note that the involvement of miRNAs is bi-directional, with stromal cells also affecting cancer cells via the release of micro-RNAs (Musumeci et al., 2011; Nouraee et al., 2013). The hypoxic conditions of the TME result in the generation of ROS. Toullec et al. (2010) demonstrated that ROS within the TME induce the expression of hypoxia inducible factor 1-α (HIF1-α) and CXC-chemokine ligand 12 by stromal fibroblasts, allowing the cells to have enhanced metabolism and migratory capabilities.
Albrengues et al. (2015) showed that there is an epigenetic switch that drives the transformation of normal fibroblasts into CAFs. Leukemia inhibitory factor, a member of the IL-6 superfamily, was shown to initiate the epigenetic switch, leading to the activation of the Janus kinase–signal transducer and activator of transcription (JAK-STAT) signaling (Albrengues et al., 2015). The continuous activation of the JAK-STAT signaling leads to CAFs promoting cancer cell invasive behavior. Several other studies have shown the importance of the JAK-STAT signaling in the maintenance of the CAF phenotype (Gao et al., 2009; Huynh et al., 2017; Tan et al., 2019). The continuous interaction between CAFs and cancer cells means that during tumor progression CAFs also change, with CAFs demonstrating heterogeneity at both phenotype and function level (Bruzzese et al., 2014; Scherz-Shouval et al., 2014).
The growth and transformation of normal epithelial cells is driven partly by CAFs (Olumi et al., 1999). Olumi et al. (1999) demonstrated that CAFs and not normal fibroblasts were responsible for the transformation as well as cancer initiation and growth of prostatic epithelial cells. Kuperwasser et al. (2004) also demonstrated that TGF-β from CAFs was necessary for tumor formation from epithelial cells, a result that was in contrast to the use of normal fibroblasts. In addition, Shekhar et al. (2001) demonstrated that normal fibroblasts inhibited tumor growth and the tumorigenic transformation of epithelial cells.
Implantation of tumor cells and CAFs together allowed enhanced tumor growth compared with co-implantation of tumor cells and normal fibroblasts (Picard et al., 1986). Several studies have shown the involvement of both cancer cell-derived and CAF-derived TGF-β and heat shock factor-1 in driving tumor cell growth and metastasis (Hawinkels et al., 2014; Scherz-Shouval et al., 2014; Senthebane et al., 2017). Using both esophageal and breast cancer cells, we demonstrated that both CAFs and cancer cells released TGF-β, which promoted the transformation of fibroblasts in addition to promoting tumor growth (Senthebane et al., 2017). Several cancer cells have been shown to express TGF-β receptors on their cell surfaces, supporting the notion that cancer cells respond to both autocrine and paracrine TGF-β signaling (Calon et al., 2014; Li et al., 2005).
Besides releasing growth factors, CAFs also influence tumor growth through the release of enzymes such as MMPs and urokinase-type plasminogen activator (Danø et al., 2005; Deryugina and Quigley, 2015; Egeblad and Werb, 2002; Martens et al., 2003; Taguchi et al., 2014).
Although further research into the role of CAFs into tumorigenesis is required, there is some evidence that CAFs induce epithelial cell transformation (Wang et al., 2018). To sustain tumor growth, new blood vessels are formed and these supply all the nutrients required by cancer cells as well as remove toxic substances from the tumor (Carmeliet and Jain, 2011).
CAFs release SDF-1, which has been shown to recruit endothelial progenitor cells, leading to formation of new blood vessels within the TME (Liu et al., 2019a; Orimo et al., 2005). CAFs are known to release several other angiogenic factors such as PDGF-C, VEGF-A, and secreted frizzled-related protein-2 (SFRP2) (De Palma et al., 2017; Liekens et al., 2010). Through the synthesis of ECM proteins and generation of ECM stiffness, CAFs influence blood vessel formation and also the flow of blood through the tumor (Egeblad et al., 2010; Senthebane et al., 2018; Twardowski et al., 2007).
Most early-stage epithelial tumors have a basal lamina that acts as a barrier between the tumor and the vascular system (Bluff et al., 2009; Bossi et al., 1995). This results in early-stage tumors lacking proper supply of nutrients and removal of toxic substances. Migration and invasion of tumor cells into the surrounding tissues induce angiogenesis as well (Bergers and Benjamin, 2003). The induction of angiogenesis is coupled to infiltration of leukocytes, proliferation and activation of fibroblasts, as well as increased deposition of ECM proteins (Dai et al., 2009; De Palma et al., 2017; Neve et al., 2014; Wang et al., 2011; Zhou et al., 2006). Depending on the tumor type, stromal components, and site in the human body, vascularization can take different forms and patterns (Bluff et al., 2009; Fukumura et al., 1997; Jubb et al., 2011; Monsky et al., 2002).
In addition, the balance of expression of anti-angiogenic factors and pro-angiogenic factors will ultimately play a key role in determining both formation and pattern of blood vessels within the tumor (Bussolino et al., 1991; Monsky et al., 2002; Morrissey et al., 2008; Murakami and Simons, 2008).
Ultimately, pro-angiogenic factors and signaling are eventually upregulated during tumor formation, leading to aberrant and dysregulated formation of blood vessels (Bergers and Benjamin, 2003; Castello et al., 2017). CAFs together with infiltrating leukocytes, macrophages, and other stromal cells such as pericytes are known to be the major sources of pro-angiogenic factors such as VEGFA, PDGFC, fibroblast growth factor 2 (FGF2), Osteopontin, and MMPs (Table 1, below) (Anderberg et al., 2009; Crawford et al., 2009; Deroanne et al., 1997; Fukumura et al., 1998; Gerber et al., 2000; Morikawa et al., 2002; Zhang et al., 2017; Zhao et al., 2014).
Several Angiogenic Factors Are Produced by Cancer-Associated Fibroblasts, Cancer-Associated Macrophages, and Cancer-Associated Neutrophils and Influence Vascularization Within the Tumor Microenvironment
CAFs, cancer-associated fibroblasts; CAMs, cancer-associated macrophages; CANs, cancer-associated neutrophils; ECM, extracellular matrix; FGF, fibroblast growth factor; MMPs, matrix metalloproteases; PDGFC, platelet-derived growth factor C; VEGFA, vascular endothelial growth factor-A.
For blood vessels to be formed there has to be the right tumor or TME elasticity and stiffness (Egeblad et al., 2010; Senthebane et al., 2017). The CAFs synthesize the TME ECM; whereas enzymes released by CAFs such as MMPs, hydroxylases, and lysyl oxidases influence both ECM synthesis and degradation and, therefore, control biophysical properties of the TME (De Palma et al., 2017; Dzobo et al., 2012, 2014, 2018a; Liu et al., 2019b; Taguchi et al., 2014; Twardowski et al., 2007). Growth factors such as PDGFC act on CAFs in an autocrine manner to induce the secretion of FGF2 as well as osteopontin (Anderberg et al., 2009; Liu et al., 2019a; Pietras et al., 2008).
As the tumor grows in size, cancer cells migrate and invade surrounding tissues. When cancer cells breach the basement membrane, they enter into circulation, travel to distant tissues and organs, and, eventually, extravasate into new tissues, a process called metastasis. It is important to note that only a few cancer cells survive the arduous journey to new tissues and organs, but once there, these cancer cells colonize and form new tumors, metastases (Chaffer and Weinberg, 2011; Gupta and Massagué, 2006). Grum-Schwensen et al. (2005) demonstrated that tumors without stromal cell-derived mts1 protein do not metastasize, demonstrating the importance of stromal cells in tumor growth and spread.
Several studies have shown that besides stromal-derived growth factors, several cytokines are also released by stromal cells and aid cancer cell metastasis (Chow and Luster, 2014; Guo and Deng, 2018; Valkenburg et al., 2018). IL-6 has received a lot of attention and has been shown to help cancer cells metastasize to the bone (Tawara et al., 2011). Chang et al. (2013) showed that IL-6 and its downstream signaling cascades such as JAK-STAT are involved in breast cancer tumorigenesis and metastasis.
The CAF-derived SDF1 has been shown to aid breast cancer cells home to the bone and aid in adaptation to this new environment (Al-Ansari et al., 2013; Zhang et al., 2013b). The release of stanniocalcin-1 (STC1) by CAFs drives colon cancer metastasis (Peña et al., 2013). TGF-β has been implicated in many cellular processes, and Yu et al. (2014) demonstrated that TGF-β can induce EMT in cancer cells. Both TGF-β and hepatocyte growth factor (HGF) have been shown to promote invasiveness in esophageal cancer (Grugan et al., 2010; Wang et al., 2016).
By releasing huge amounts of MMPs as well as synthesizing the ECM, CAFs can remodel the TME and allow cancer cells to migrate and invade surrounding tissues. Increase in collagen levels within the TME has been shown to promote cancer cell invasiveness (Provenzano et al., 2008; Vellinga et al., 2016). Degradation of the ECM by CAF-derived MMPs can create highways through which cancer cells can migrate to other tissues and organs (Glentis et al., 2017; Senthebane et al., 2018). In addition, ECM stiffening is enhanced via activation of YAP1 in CAFs and has been shown to allow cancer cell invasion and formation of tumor blood vessels (Calvo et al., 2013).
Goetz et al. (2011) demonstrated that CAF-driven biomechanical remodeling of the TME can allow migration and invasion of cancer cells. Once cancer cells reach their new environment, it has to be remodeled to suit their needs or the cancer cells adapt to the new conditions. As postulated by Paget in the “seed and soil” theory, the new microenvironment must allow cancer cells to flourish (Paget, 1889).
Kaplan et al. (2005) suggested that a premetastatic environment must be rich in ECM proteins such as fibroblast-derived fibronectin. In addition, stromal cell-derived factors including TGF-β and SDF1 within the premetastatic regions may act as attractants to cancer cells (Calon et al., 2012; Kaplan et al., 2005). Resident fibroblasts within the premetastatic regions promote blood vessel formation, allowing tumors to grow (Calon et al., 2012; O'Connell et al., 2011). It is plausible to suggest that these fibroblasts and any other stromal cells associated with metastatic cancer cells are resident cells within the colonized regions.
The Role of CAFs in Therapy Resistance
There are two possible ways through which cancer cells can be resistant to therapy. First, therapy resistance can be intrinsic. The expression of several transporter proteins such as the ABC proteins and other genetic alterations can influence drug uptake and export at the cellular levels. Second, tumors are always evolving and this can result in previously responsive tumors to become irresponsive. The TME has emerged as a contributor to therapy resistance via the actions of stromal cells and the ECM. Excessive synthesis of the ECM by CAFs can limit drugs' access to cancer cells and other stromal cells. The ECM around cancer cells can form a hindrance to drugs and can limit blood vessel formation within the tumor (Olive et al., 2009; Zhao et al., 2018).
The binding of cancer cells to CAF-derived ECM proteins such as fibronectin, a process called cell-adhesion-mediated drug resistance, also aids cancer cells in avoiding drugs, thus causing drug resistance (Damiano et al., 2001; Hazlehurst et al., 2000). The versatile IL-6 cytokine has been implicated in resistance to chemotherapy in lung cancer (Shintani et al., 2016). Although it is understandable that cancer cells can develop therapy resistance, CAFs have also been reported to be resistant to several drugs, including gemcitabine in pancreatic cancer (Richards et al., 2017). Communication between CAFs and cancer cells also includes exosomes, which transport several factors and miRNAs from CAFs to cancer cells (Richards et al., 2017).
Most chemotherapeutic drugs target rapidly growing cancer cells, but they cannot effectively clear all cancer cells during treatment. In addition, chemotherapy induces DNA damage in stromal cells, resulting in the release of several factors that promote cancer cell survival (Huber et al., 2015). Further, several studies demonstrated that chemotherapy can induce fibroblasts to become CAF-like cells that promote stem cell behavior in cancer cells (Peiris-Pagès et al., 2015a, 2015b). Radiotherapy is known to induce the synthesis of large amounts of ECM proteins, resulting in the survival of cancer cells (Barker et al., 2015; Eke and Cordes, 2011; Erkan et al., 2007). When irradiated, stromal cells can increase the secretion of factors involved in chemoresistance (Chargari et al., 2013).
The EMT process is used by cancer cells to transform and reduce the expression of many cellular membrane proteins such as drug transporters (Zheng et al., 2015). The net effect of reduced drug transporter expression is reduced drug intake, ultimately leading to therapy resistance. The EMT is a process that is activated by several signaling pathways and occurs in normal events such as embryonic development.
Unfortunately, it also occurs during neoplastic conversion of cells as well as fibrosis (Arumugam et al., 2009; Hotz et al., 2007). Cells undergoing EMT gradually lose epithelial cell junction proteins and begin to synthesize more vimentin, becoming more migratory, invasive, and chemoresistant (Arumugam et al., 2009; Hotz et al., 2007). Several studies have shown enhanced vimentin, Snail, and ZEB1 expression in cells that are chemoresistant (Guaita et al., 2002; Taube et al., 2010; Wellner et al., 2009). Zheng et al. (2015) demonstrated that although EMT is not necessary for metastasis, it induces the development of chemoresistance in pancreatic cancer.
In pancreatic cancer, resistance to gemcitabine is associated with EMT (Vega et al., 2004; Wellner et al., 2009; Zhang et al., 2010a). Poor survival of pancreatic patients has been associated with an increased EMT program that causes an impaired response to chemotherapy (Hidalgo, 2010; Kleger et al., 2014). Even more sinister is the seemingly ability of CAFs to uptake and accumulate high concentrations of drugs within the TME, resulting in a reduced amount of drugs eventually reaching cancer cells (Hessmann et al., 2018). Chemotherapy can kill cancer cells via production of ROS. The CAFs have been shown to abrogate ROS production and therefore prevent death of cancer cells when exposed to chemotherapy (Cheteh et al., 2017).
Several survival pathways such as MEK-ERK and PI3K-Akt are known to be activated in cancer cells via the action of CAF-derived HGF and cause cancer cell resistance to RAF inhibitors (Straussman et al., 2012). In elaborate experiments, Senthebane et al. (2017) demonstrated that CAF-derived TGF-β plays a role in the resistance of breast and esophageal cancer cells to paclitaxel and cisplatin. Thus, the CAF secretome plays a huge role in the development of therapy resistance in cancer cells.
The presence of cancer stem cells (CSCs) within the TME is still debatable, but several studies have shown that these stem-like cells are able to resist therapies (Codd et al., 2018; Dawood et al., 2014; Nassar and Blanpain, 2016; Steinbichler et al., 2018; Yu et al., 2007a). Several studies have demonstrated that CAFs subsets express drug transporter proteins such as ABC transporters and also provide a protective environment to CSCs within the TME (Begicevic and Falasca, 2017; Domenichini et al., 2019; Su et al., 2018). CAF-derived cytokines such as IL-6 are responsible for maintenance of the CSC phenotype (Ebbing et al., 2019).
Microvesicles from CAF also deliver micro-RNAs to generate CSCs in breast cancer (Sansone et al., 2017). Overall, studies have demonstrated that CAFs or their subsets promote therapy resistance in cancer cells via various mechanisms from accumulating large amounts of drugs, release of protein factors, and mi-RNAs-laden exosomes to providing physical barriers to therapy. The combined targeting of these CAFs or their subsets together with cancer cells may offer durable treatment solutions for many cancers.
CAFs and Other Stromal Cell Interactions
The interaction between TME components is through cell-to-cell adhesions, release of growth factors, and cytokines and indirectly through exosomes. In addition, cells interact with the ECM via surface receptors such as integrins. Crosstalk between stromal cells is inevitable given the compact nature of most solid tumors. The most abundant immune cells within the TME are the macrophages, referred to as cancer-associated macrophages (CAMs) or tumor-associated macrophages (TAMs).
As reported by Comito et al. (2014), CAFs and TAMs work together to promote tumor progression in prostate cancer. Communication between the two types of cells occurs via the release of CXCL14 by CAFs, resulting in the recruitment of TAMs to the tumor site and subsequent differentiation (Comito et al., 2014). In turn, M2 TAMs are known to activate CAFs and therefore further promote tumorigenic growth. Hashimoto et al. (2016) demonstrated that CAFs derived from bone marrow MSCs promote macrophage invasiveness and transformation and, in turn, the resulting TAMs can prompt the proliferation of CAFs. By working together, these CAFs and TAMs can promote tumorigenic growth of neuroblastoma (Hashimoto et al., 2016).
Myeloid cells within tumors can increase the expression of S100A8 in response to IL-6 released by CAFs in colon cancer (Kim et al., 2012). Myeloid cells within tumors can also differentiate into suppressor cells that are linked to suppression of immunity (Kim et al., 2012). For example, circulating myeloid-derived suppressor cells are recruited to tumors via the release of FAP by CAFs and these cause the suppression of immunity in hepatic cancer (Yang et al., 2016).
The CAFs are known to express the FAS ligand and this is known to cause apoptotic death of CD8-positive T cells (Lakins et al., 2018). Further, CAFs express programmed cell death 1 ligand 2 (PD-L2), which causes the functional inactivation of T cells (Lakins et al., 2018). Our study and several others have shown that CAFs secrete huge amounts of TGF-β, which has been suggested to cause immunosuppression and can induce transformation of immune cells into immune-suppressing cells (Flavell et al., 2010; Gutcher et al., 2011; Senthebane et al., 2017). Recently, Mariathasan et al. (2018) provided further evidence of the involvement of TGF-β in abrogating antitumor immunity in bladder cancer. The CAFs attract and sequester CD8-positive T cells and this prevents T cells from binding to and killing cancer cells (Ene–Obong et al., 2013).
The net effect of all this is the suppression of anti-tumor activity of immune cells. The removal of FAP-positive CAFs was shown to reactivate the anti-tumor activity of immune cells of T cells (Kraman et al., 2010). Overall, CAFs are more likely to promote tumorigenic growth through their effect on immune cells within the TME. Several growth factors are known to be sequestered by the ECM and these include VEGFA and TGF-β. Through production of matrix metalloproteases (MMPs), CAFs can degrade the ECM and cause enhanced levels of free growth factors within the TME (Bonnans et al., 2014). Thus, through recruiting endothelial cells in addition to releasing VEGFA, CAFs promote tumor vascularization (Bonnans et al., 2014). The interaction of CAFs and other cells, therefore, creates a tumor environment that promotes immune evasion, inflammation, and vascularization.
As reported by Senthebane et al. (2017), CAFs and other TME components can also display anti-tumorigenic effects, especially during the early stages of tumor development. Our data revealed that normal fibroblast-derived ECM limits cancer cell proliferation and migration compared with an ECM from fibroblasts co-cultured with cancer cells (Senthebane et al., 2018). Further evidence of the involvement of CAFs or their normal counterparts in anti-tumorigenic behaviour comes from studies of breast cancer. Brechbuhl et al. (2017) reported two subsets of CAFs in breast cancer, with one subset expressing CD146 and conferring sensitivity to tamoxifen, whereas the CD146- subset confers tamoxifen resistance. The removal of CAFs expressing α-SMA from pancreatic ductal adenocarcinoma (PDAC) results in the acceleration of cancer and reduced survival (Özdemir et al., 2014; Rhim et al., 2014).
With this in mind, novel therapeutic strategies aimed at CAFs have to be selective and cognizant of the CAFs' phenotypic and functional heterogeneity. Mere targeting of CAFs for removal based on presumed pro-tumorigenic behavior will be detrimental and can lead to fatal disease. The CAF subsets must be identified and only targeted therapy should be used. In addition, it might be helpful to re-educate or re-direct the pro-tumorigenic CAFs into anti-tumorigenic CAFs. To be able to do this, it is important that specific markers for anti-tumorigenic and pro-tumorigenic CAFs be identified to distinguish the two.
Advances in Therapeutic Targeting of CAFs
CAF-directed therapies
It has long been recognized that manipulation of CAFs' functions, especially the tumorigenic promotion part, have therapeutic value in cancer treatment. In theory, targeting CAFs appear easy since they are the major stromal components of the tumor. In addition, CAFs have been described as genetically stable, making them a better target than cancer cells in cancer immunotherapy (Ostermann et al., 2008). In practice, however, major challenges need to be overcome before this promising avenue is utilized in cancer treatment.
First, targeting CAFs will have a negative effect on normal tissue and other components of the TME. For example, the depletion of CAFs can result in less ECM production, creating highways through which cancer cells escape and metastasize to other tissues and organs. Second, the removal of CAFs itself suffers from the lack of specific markers that can be used to achieve that. Third, and probably most challenging is the existence of CAFs subsets displaying phenotypic and functional heterogeneity. To overcome this requires the identification of more than one specific markers of each subset that can be used for targeting that subset.
Several approaches have been devised to target CAFs within the TME. First, the activation and trans-differentiation of stromal cells into CAFs can be targeted and inhibited. Since CAFs influence both tumor cells and stromal cells via synthesis and secretion of growth factors and cytokines, inhibition of this synthesis and secretion is under intense scrutiny, with the aim of inhibiting CAF function. Second, CAFs' normalization through the use of several molecules has been tried.
For example, all-trans-retinoic acid (ATRA) can be used to induce quiescence in CAFs or activated fibroblasts/stromal cells, preventing aberrant secretion of growth factors and cytokines. Although CAFs are pro-tumorigenic, they can be used as carriers of drugs to kill cancer cells. Together with other cells such as CAMs, MSCs, and pericytes, CAFs can carry anti-cancer viruses and apoptosis-inducing ligands. Several studies, including our recent publication, have demonstrated that MSCs can be transformed into CAFs (Borriello et al., 2017; Chan et al., 2019; Ridge et al., 2018; Senthebane et al., 2017; Tan et al., 2019). Senthebane et al. (2018) demonstrated the influence of the ECM in tumor growth and chemoresistance.
Importantly, the presence of ECM proteins, including type I collagen and fibronectin, was shown to affect drug delivery to cancer cells, with the knockdown of both ECM proteins showing increased drug-induced cancer cell death (Senthebane et al., 2018). Previous studies implicated signaling pathways such as the MEK-ERK and MMPs in regulating ECM synthesis (Dzobo et al., 2012, 2014). The ablation of stromal ECM can, therefore, be used to increase cancer cell sensitivity to anti-cancer drugs. Direct targeting of CAFs or their subsets is still challenging, with difficulties in the identification of markers to be used to identify the CAFs. Currently, immunotherapy, use of immunotoxins, and DNA vaccines can be used to target CAFs, with the duly still out on the effectiveness of such therapy.
Senthebane et al. (2017) recently utilized both vimentin and α-SMA as markers to identify MSC-derived CAFs. Other studies have shown that α-SMA only identifies a specific subset of CAFs (Choi et al., 2013; Ding et al., 2014; Özdemir et al., 2014). Özdemir et al. (2014) described that the removal of α-SMA-positive CAFs in pancreatic ductal adenocarcinoma (PDAC) was linked to decreased angiogenesis. The same authors observed that angiogenesis decreased whereas there was increased hypoxia within solid tumors leading to increased EMT and the presence of CSCs (Özdemir et al., 2014). Several lung and colon cancer animal models have shown that removal of FAP from within solid tumors is associated with decreased tumor growth (Santos et al., 2009).
Santos et al. (2009) demonstrated that a preclinical FAP inhibitor, PT630, can inhibit the growth of tumors as well as stromagenesis in lung and colon cancers. Similarly, the removal of FAP in PDAC also reduced tumor growth (Feig et al., 2013). Ostermann et al. (2008) developed a monoclonal antibody against FAP that they conjugated to maytansinoid and this treatment has been used in pancreatic, head, and neck cancers where it has shown its effectiveness.
In addition, extensive experiments by Loeffler et al. (2006) using a mouse DNA vaccine against FAP resulted in ablation of CAFs and enhanced drug uptake by tumors. The DNA vaccine induced a CD8+ T cells to kill a large part of the CAFs expressing FAP. Further evidence for the utility of the DNA vaccine against FAP-positive CAFs was provided by Reisfeld (Reisfeld, 2007). The utility of chimeric antigen receptor T (CAR T) cell treatment for solid tumors is still to be proven.
Several reports, however, show that targeting FAP-positive CAFs using CAR T cells results in antitumor activity (Kakarla et al., 2013; Lo et al., 2015; Wang et al., 2014). However, CAFs are not the only cells expressing FAP. Bone marrow-derived MSCs are also known to express FAP (Chung et al., 2014). Thus, it is possible that anti-tumor CAR T cells will also target other cells. Although reports show that CAR T cells targeting FAP-positive CAFs are able to kill cancer cells in lung and pancreatic cancers (Lo et al., 2015), Roberts et al. (2013) demonstrated that CAR T cells killed bone marrow-derived stem cells and can cause cachexia and anemia.
Increased knowledge and analytical tools have enabled scientists to identify new and better surface markers for CAFs. For example, Su et al. (2018) were able to characterize tumorigenic CAF subsets based on the expression of CD10 and GPR77. Used together with FAP and α-SMA, these new surface markers are likely to define specific CAF subsets, leading to their killing.
The same authors reported reduced cancer cell stemness when GPR77 was blocked (Su et al., 2018). Several strategies are being developed to target cellular sources of CAFs. Senthebane et al. (2017) demonstrated that it is possible that MSCs are transformed into CAFs through their interaction with cancer cells. Thus, targeting MSCs may prevent the accumulation of CAFs in tumors, reducing pro-tumorigenic cells within the tumor. The removal of pro-tumorigenic support is likely to reduce tumor growth and metastasis. Several clinical trials have been undertaken to target CAFs sources with the duly still out on their effectiveness.
The re-education of activated CAFs has been suggested as a strategy to direct CAFs from being pro-tumorigenic to anti-tumorigenic. The inactivation of CAFs can induce quiescence, with the resulting CAFs or fibroblasts dividing slowly and releasing normal levels of growth factors and other factors. Akin to quiescent satellite cells in muscle, these re-educated CAFs can attain a tumor-suppressive phenotype. The induction of quiescence in pancreatic stellate cells in PDAC through the use of ATRA reverts the activated cells to normal fibroblasts and arrests tumor growth (Froeling et al., 2011). The mechanism of action of ATRA involves inhibition of the versatile Wnt-β-catenin signaling cascade (Froeling et al., 2011).
Inactivated pancreatic stellate cells also release sequestered CD8+ T cells, allowing infiltration into PDAC and resulting in tumor growth arrest (Ene–Obong et al., 2013). Carapuca et al. (2016) showed that coupling ATRA, a vitamin A analogue, and gemcitabine can be effective at treating PDAC in animal models through inhibition of several pathways, including Wnt-β-catenin and Hedgehog signaling. Sherman et al. (2014) demonstrated that global reprogramming of the stroma through the use of calcipotriol, a vitamin D receptor ligand, and gemcitabine resulted in reduced inflammation and ECM synthesis in PDAC tumors. Reduced ECM synthesis allowed better delivery of gemcitabine into the tumor, leading to tumor regression (Sherman et al., 2014).
Both ATRA and calcipotriol are at the preclinical stage of investigations. Thus, depending on the tumor type, different strategies from ablation of CAFs to normalization of activated CAFs may be undertaken to achieve better cancer treatment. This provides a new avenue of cancer treatment that focuses not just on cancer cells but on the stromal component as well. Combinations of strategies focused on cancer cells and stromal cells may offer a durable cure for cancer treatment.
Targeting signaling pathways activated in CAFs and their downstream effectors
One of the remaining challenges to CAFs' ablation and normalization is the huge heterogeneity observed in many tumors, meaning some CAFs subsets will remain unaltered by treatment. Instead of targeting the CAFs, several approaches have been developed to target signaling pathways activated in CAFs and factors released by tumor and stromal cells. Tumor cells and stromal cells release huge amounts of factors during tumor initiation and progression (Senthebane et al., 2017, 2018; Sullivan et al., 2010). Senthebane et al. (2017) demonstrated that TGF-β released by tumor cells and transforming MSCs can be involved in the transformation of stromal cells into potential CAFs. The same study also showed that once transformed, activated CAFs release increased levels of the same growth factor, TGF-β, which is involved in tumor growth and development of chemoresistance (Senthebane et al., 2017).
One signaling cascade that has been studied in detail in different cellular processes is the IL-6-JAK-STAT pathway (Giannoni et al., 2010; Sansone and Bromberg, 2012; Steyn et al., 2019). Sanz-Moreno et al. (2011) showed that Oncostatin M, a member of the IL-6 superfamily, activates STAT3 signaling and drives ECM remodeling. ECM remodeling allows cancer cells to invade surrounding tissues, escape, and metastasize to other sites (Sanz-Moreno et al., 2011). A detailed description of inhibition of the IL-6-JAK-STAT signaling pathway as well as the TGF-β signaling is given next.
Pietras et al. (2008) demonstrated that the reduction of FGF2 and fibroblasts growth factor-7 (FGF7) in animal models of cervical cancer via Imatinib-mediated inhibition of PDGF receptor slowed cancer cell division and disrupted angiogenesis. The authors showed that targeting CAFs can act in complement to conventional therapeutic strategies and improve the management of cancers that are difficult to cure. Research by Anderberg et al. (2009) further showed that PDGF-CC induces the expression of osteopontin in CAFs, leading to tumor growth acceleration.
The chemokine SDF1, exclusively produced by FAP-positive CAFs, binds to its receptor CXCR4, resulting in the suppression of immunity within the TME through the prevention of CD8+ T cell infiltration (Fearon, 2015; Feig et al., 2013; Kraman et al., 2010). Several inhibitors of SDF1-CXCR4 interactions have been developed and these can reactivate the anti-tumor immunity by enhancing the infiltration of CD8+ T cells into the TME (Feig et al., 2013). Inhibition of SDF1 and CXCR4 interactions include the use of inhibitors such as AMD3100 and may enhance the anti-tumor effects of monoclonal antibodies against PDI/PDL1 in pancreatic duct adenocarcinoma (Feig et al., 2013).
Another strategy under consideration to control CAF-derived factors includes the inhibition of protein synthesis. The inhibition of protein synthesis through the use of SOM230, an inhibitor of protein synthesis in α-SMA-positive CAFs, resulted in reduced levels of several molecules (Duluc et al., 2015). Indeed, the inhibition of protein synthesis in CAFs led to less ability of cancer cells to resist chemotherapy in a PDAC model (Duluc et al., 2015).
Targeting the IL-6-JAK-STAT signaling in cancer
It has been shown that the IL-6-JAK-STAT3 is abnormally activated in several cancers, with enhanced activation of the pathway linked to poor patients' survival (Chen et al., 2013b; Kusaba et al., 2006; Macha et al., 2011). Within the TME, the IL-6-JAK-STAT3 signaling has been shown to be responsible for cancer cell invasive and metastatic behavior. In addition, the IL-6-JAK-STAT3 signaling represses immunological response to tumors. Anti-cancer agents or inhibitors against members of the IL-6-JAK-STAT3 have been approved by the FDA and the EMA, whereas others are still under investigation.
Several pathological conditions display high levels of IL-6 and increased activation of the IL-6-JAK-STA3, including rheumatoid arthritis as well as many cancers (Kumari et al., 2016; Masjedi et al., 2018). Mutations in genes of several JAK-STAT pathway members activate the pathway in many neoplasms. Increased IL-6 in circulation and within the TME is a result of the JAK-STAT pathway being constitutively activated (Kusaba et al., 2006; Taniguchi and Karin, 2014; Wang et al., 2013).
Besides CAFs, multiple other cells have been shown to produce IL-6 within the TME, from pericytes, immune cells, and even tumor cells (Bournazou and Bromberg, 2013; Nagasaki et al., 2014; Nozawa et al., 2006). The activation of STAT3 by IL-6 in tumor cells results in the expression of proliferation and survival genes, including cyclin D1 and BCL2-like protein 1 (BCL-xL), respectively (Fisher et al., 2014). Since both CAFs and tumor cells show activation of IL-6-JAK-STAT signaling, the description given next and the targeting of the IL-6-JAK-STAT signaling can therefore be applied to both CAFs and tumor cells to achieve durable cancer treatment. IL-6 activated STAT3 can induce VEGF, MMPs, and TGF-β expression, promoting angiogenesis, invasion, and development of therapy resistance, respectively (Fisher et al., 2014; Yu and Jove, 2004; Yu et al., 2009).
Besides direct effects on cancer cells, IL-6 and JAK-STAT signaling influences the behavior of stromal cells, including CAFs, CAMs, TANs, effector T cells, and killer cells (Harris et al., 2007; Iwata-Kajihara et al., 2011; Kortylewski et al., 2005; Kujawski et al., 2010; Lee et al., 2010; Yu et al., 2007b). By affecting both CAFs and immune cells, the IL-6-JAK-STAT signaling contributes to the suppression of the immune response in the TME.
The importance of IL-6 in TME is underscored by its various effects on tumor cell and stromal cell survival, proliferation, and invasiveness (Kortylewski et al., 2005; Lederle et al., 2011). Two IL-6 signaling pathways are known: the classical IL-6 signaling pathway, which involves IL-6 binding to the IL-6 receptor on the cell surface and interacts with transmembrane protein gp130; the trans-signaling pathway whereby IL-6 binds to a soluble form of the IL-6 receptor and then interacts with gp130. Importantly, although similarities exist in the ways in which both signaling pathways regulate cell behavior, the trans-signaling pathway controls the recruitment and activation of stromal cells (Campbell et al., 2014; Hunter and Jones, 2015). Activation of the IL-6 signaling results in activation of mostly JAK1 and JAK2, which then phosphorylate members of the STATs family (Johnson et al., 2018; Steyn et al., 2019).
Of the STATs proteins, STAT3 is the most studied and has been linked with tumor progression and suppression of the immune system (Bromberg, 2002; Yu et al., 2009). Activation of STATs proteins is regulated by PIAS proteins, suppressor of cytokine signaling proteins, phosphatases, and miRNAs (Dorritie et al., 2014; Kim et al., 2010; Sekine et al., 2006; Zhang et al., 2007).
The IL-6 levels are increased in cancers, including breast, lung, colorectal, esophageal, ovarian, and prostate cancers (Chen et al., 2013a; Chung and Chang, 2003; Culig and Puhr, 2012; Dethlefsen et al., 2013; Macciò and Madeddu, 2013). Several preclinical studies utilizing models and patient samples have demonstrated the critical roles of circulating IL-6 in tumor development (Becker et al., 2004; Lesina et al., 2011; Sansone et al., 2007). For example, IL-6 enhances CSC proliferation in breast cancer (Sansone et al., 2007). In addition, IL-6 is known to promote EMT and metastasis in breast cancer (Chang et al., 2013; Sullivan et al., 2009). Consequently, IL-6 levels in circulation have been used as prognostic indicators in patients' survival and can predict response to therapy (Gao et al., 2016; Knupfer and Preiss, 2010; Maccio and Madeddu, 2013; Sanguinete et al., 2017).
Several studies utilizing models and patients' samples have demonstrated the critical role played by CAF- and tumor-derived IL-6 cancers, including breast, colorectal, esophageal, lung, and skin cancers (Gao et al., 2007; Grivennikov et al., 2009; Lederle et al., 2011; Sansone et al., 2007). Aberrant levels of STAT3 activation have been shown in many cancers (Frank, 2007; Roeser et al., 2015). Besides being involved in tumor progression, several studies have shown that STAT3 can promote therapy resistance (Sen et al., 2012; Spitzner et al., 2014).
Several strategies have been developed to target the IL-6-JAK-STAT signaling pathway (Fig. 3). Siltuximab, for example, targets IL-6 directly and Tocilizumab targets the IL-6 receptor. Siltuximab has shown efficacy against several solid tumors, including lung and prostate cancers (Cavarretta et al., 2008; Song et al., 2014). Siltuximab have been shown to reduce the levels of STAT3 and other signaling cascades such as MEK-ERK in tumors (Fizazi et al., 2012; Karkera et al., 2011). Tocilizumab is used in patients with rheumatoid arthritis and has efficacy against colorectal and pancreatic cancers (Dijkgraaf et al., 2015; Grivennikov et al., 2009).
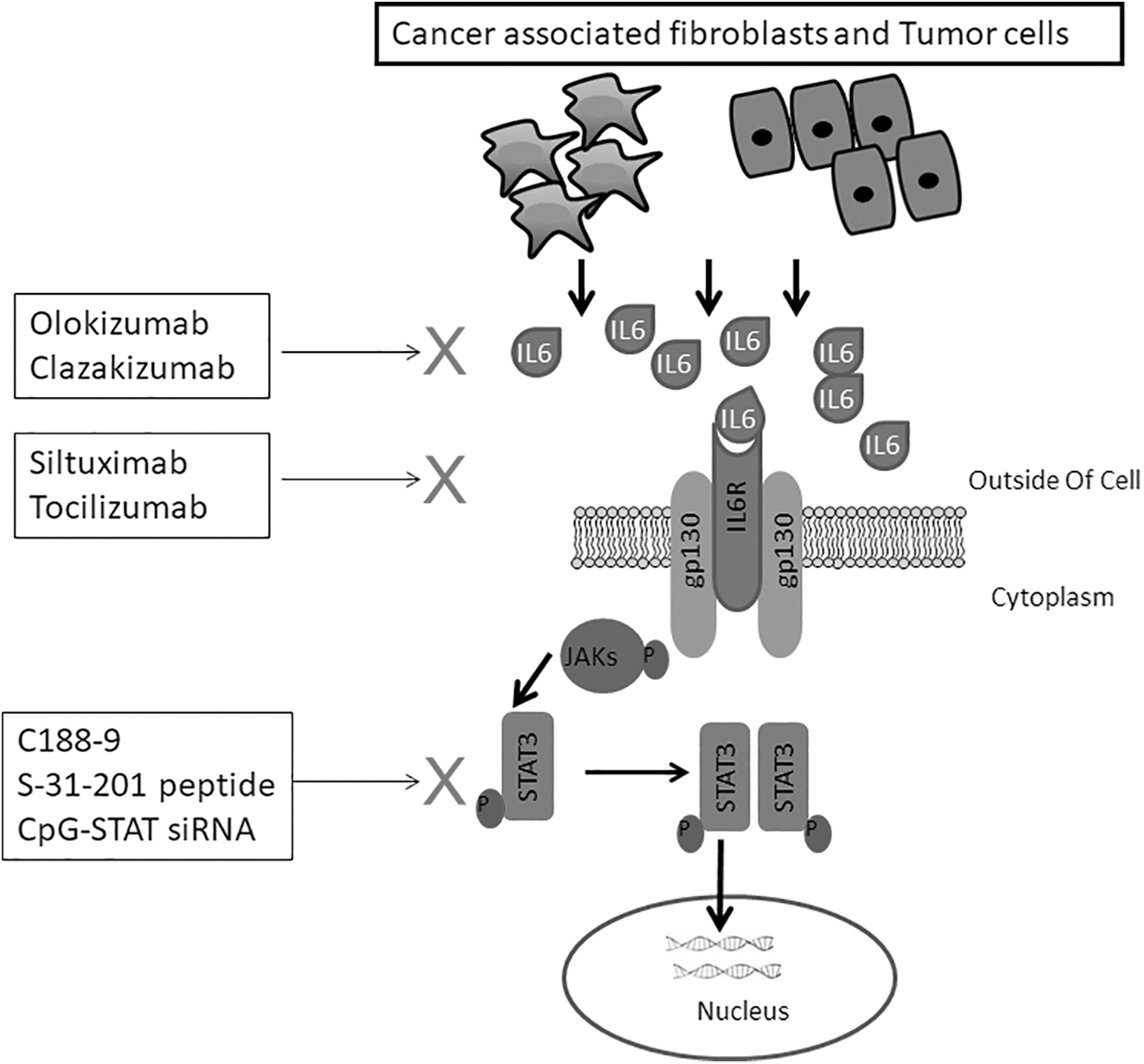
Some of the strategies targeting both CAF- and tumor cell-derived IL-6-JAK-STAT signaling pathways are indicated. JAK, Janus kinase; STAT, signal transducer and activator of transcription; X, blocking of signaling step.
Inhibitors of JAKs include Pacritinib, Ruxolitinib, and Tofacitinib and these have shown varied efficacy against cancers such as colitis, myelofibrosis, liver, pancreatic, and ovarian cancers (Hedvat et al., 2009; Komrokji et al., 2015; Neubauer et al., 1998; Sandborn et al., 2017; Tavallai et al., 2016; Vannucchi et al., 2015). The STAT3 inhibitors demonstrated antitumor activities and promoted apoptosis against several cancer cells (Chen et al., 2007; Fuh et al., 2009; Pan et al., 2013; Schust et al., 2006; Siddiquee et al., 2007; Zhang et al., 2010b).
Targeting the CAF-derived TGF-β signaling pathway in cancer
Several reports document the lack of cancer patients' response to therapy being associated with TGF-β signaling in CAFs and CAMs (Brunen et al., 2013; Colak and Ten Dijke, 2017; Ganesh and Massague, 2018; Hao et al., 2019; Jakowlew, 2006; Mariathasan et al., 2018; Reiss, 1999). In normal cells and during early stages of tumor development, the TGF-β signaling displays anti-tumor functions, with reports of cancer cell cycle arrest and induction of apoptosis (Colak and Ten Dijke, 2017; Gu and Feng, 2018; Senthebane et al., 2017).
Our study demonstrated that as tumors develop, TGF-β from both stromal cells and tumor cells participates in the transformation of normal fibroblasts and MSCs into CAFs and thus contributes to further tumor development, metastasis, and therapy resistance (Fig. 4) (Bussard et al., 2016; Castells et al., 2012; Senthebane et al., 2017). Thus, depending on the stage of tumor development, TGF-β signaling can have both tumor-promoting and tumor-suppressive functions. Targeting this pleiotropic signaling cascade is therefore difficult, with further research required to determine the specific members of the pathway to target as well as the right doses. Importantly, the effects of inhibition of this signaling cascade on both cancer cells and stromal components must be further investigated. The elucidation of possible biomarkers that can point to the usefulness of potential inhibitors during and after treatment must be determined.
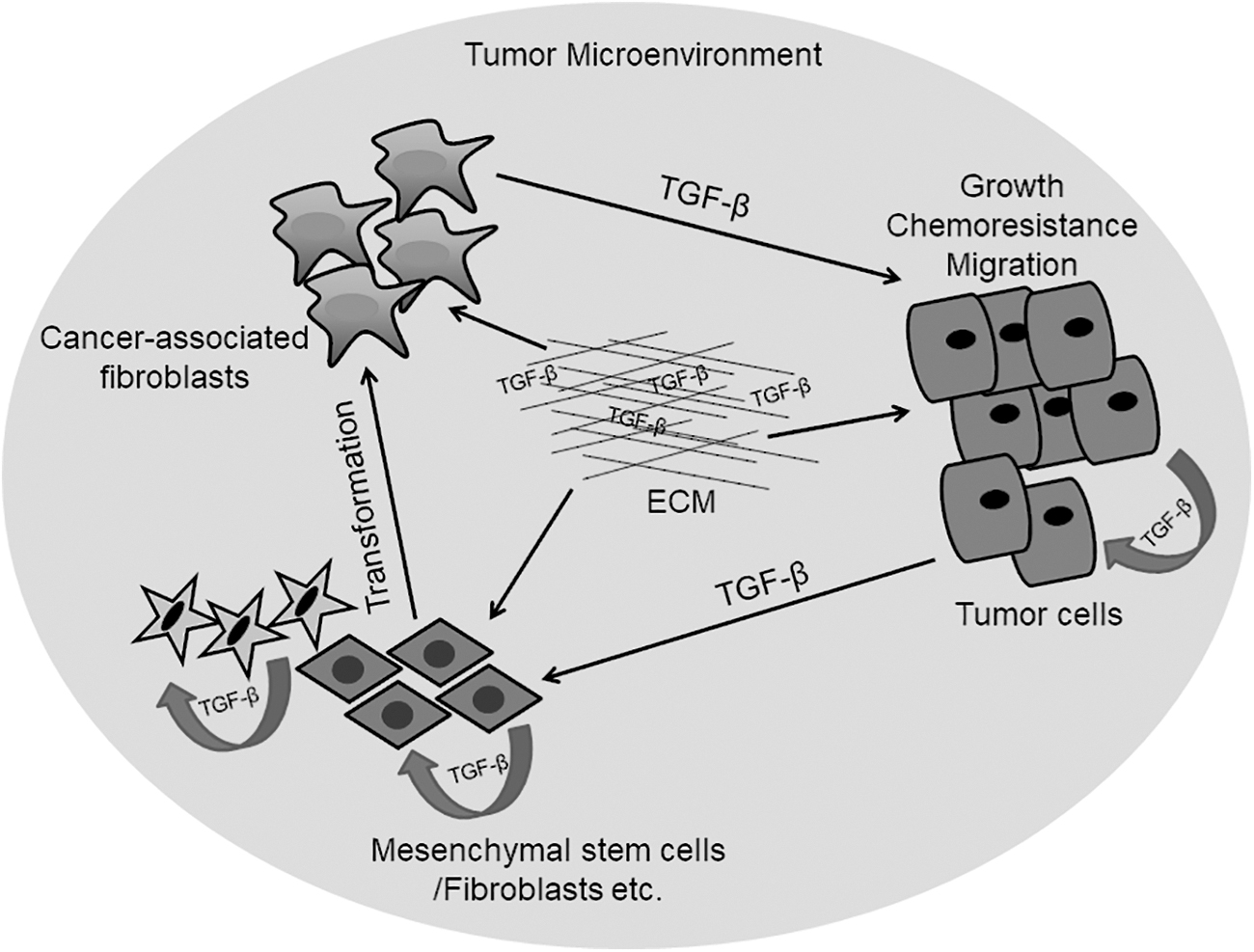
Both tumor and stromal cells secrete TGF-β into the TM milieu. TGF-β and other growth factors can be sequestered by the ECM. Increased TGF-β within the TM leads to transformation of MSCs and fibroblasts into CAFs. Increased CAF- and tumor-derived TGF-β levels cause increased tumor growth, chemoresistance, and metastasis. Figure adapted from Senthebane et al. (2017).
In their elaborate study, Mariathasan et al. (2018) demonstrated that a combination of TGF-β signaling blockade and anti-PD-L1 repressed TGF-β signaling in stromal cells, leading to enhanced tumor suppression. Importantly, the authors demonstrated the need to combine treatment strategies to attain durable cure in cancer. Several drugs targeting TGF-β are under trial and these include fresolimumab (NCT02581787).
Targeting CAF-derived ECM proteins and associated signaling
The CAFs are the principal cells responsible for synthesizing the ECM (Barbazan and Matic Vignjevic, 2019; Erdogan and Webb, 2017; Houthuijzen and Jonkers, 2018; Khan et al., 2018; Liu et al., 2019b). In normal tissue, fibrillar collagens, fibronectin, hyaluronan, and tenascin C are deposited in large amounts to make the ECM. Increased synthesis of ECM proteins results in desmoplastic reactions and these are known to drive tumor growth (Ronnov-Jessen et al., 1996). Besides inducing desmoplastic reactions, increased ECM synthesis creates a barrier for the delivery of drugs to cancer cells (Laklai et al., 2016; Senthebane et al., 2017). In detailed in vitro experiments, we demonstrated that the presence of an ECM increased the resistance of breast and esophageal cancer cells to several chemotherapeutic agents (Senthebane et al., 2018).
Although it is plausible that a reduction in ECM synthesis in CAFs and other stromal cells such as macrophages is an appealing strategy, improper ECM synthesis can create “highways” through which cancer cells may metastasize (Jabłońska-Trypuć et al., 2016; Senthebane et al., 2017, 2018). Increased amounts of both collagen and hyaluronan in tumors can result in compressed vascular networks, inadvertently reducing the flow of drugs to cancer cells (Chauhan et al., 2013; Dzobo et al., 2014). Targeting both collagen and hyaluronan production by both CAFs and macrophages can allow drugs to reach cancer cells (Chauhan et al., 2013). The inhibition of collagen through the use of halofuginone has been shown to reduce desmoplasia, impacting tumor progression (Juarez et al., 2012).
Enzymatic degradation of ECM proteins, such as collagen via MMPs occurs during ECM remodeling. The use of MMPs as an anti-cancer therapy has largely failed to materialize (Vandenbroucke and Libert, 2014). For a start, there is so much overlap in MMP activities and indiscriminate degradation of ECM proteins can have a negative effect during treatment (Vandenbroucke and Libert, 2014).
Currently, several MMPs inhibitors are under investigation in many cancers (Chiappori et al., 2007). Deciphering the exact MMPs activities and their specific targets can aid in the development of novel use of MMPs in cancer treatment (Coussens et al., 2002; Dzobo et al., 2012; Wojtowicz-Praga et al., 1997). Hyaluronan can be depleted through the use of recombinant hyaluronidase enzyme to allow drugs to reach cancer cells (Jacobetz et al., 2013; Provenzano et al., 2012). Targeting hyaluronic acid via the use of PEGPH20 is still under investigation (NCT01453153). New clinical trial data (HALO-109-301) show that when used together with gemcitabine and nab-paclitaxel in patients with advanced pancreatic cancer, PEGPH20 is promising (Doherty et al., 2018).
Caution, however, must be heeded as the resulting expansion of the vascular system can result in invasion and metastasis of cancer cells. A combination of the removal of ECM proteins during treatment can enhance patients' survival, demonstrated in PDAC by Hingorani et al. (2016). In vitro work by Senthebane et al. (2018) demonstrated that the removal of both collagen and fibronectin from ECM resulted in increased apoptosis in esophageal cancer cells. Antibodies have been used to target fibronectin to reduce vascularization and tenascin C to prevent metastasis (Ebbinghaus et al., 2004; Reardon et al., 2006). Targeting Tenascin C together with chemotherapy improves the survival of glioma patients (Reardon et al., 2006). The inhibition of signaling pathways perturbed in TME such as the IL-6-JAK-STAT and sonic hedgehog cascade can aid in reducing CAFs and their secreted factors, resulting in better therapy response (Olive et al., 2009).
For example, IPI-926, an inhibitor of the Hedgehog pathway, together with Gemcitabine has been shown to influence resistance to chemotherapy in PDAC (Olive et al., 2009). Currently, most stromal-directed therapy is still in its infancy and removal of any stromal elements must be studied carefully. Stromal cells and their products such as the ECM are needed for normal tissue architecture and homeostasis (Dzobo et al., 2012; Senthebane et al., 2017, 2018; Sherman et al., 2014). Any stromal-directed therapy will have to work in combination with tumor-directed therapy to achieve durable cancer treatment.
Targeting CAF- or stromal-induced angiogenesis
Given the diverse cells and mechanisms through which tumor vasculature is induced and develops, the inhibition of tumor angiogenesis is challenging. Although angiogenesis inhibition has been successful in some cases, partial to total failure has also been reported in some studies (Bergers and Hanahan, 2008; Ferrara and Adamis, 2016; Ferrara, 2016; Jayson et al., 2016; Okaji et al., 2008; Patrikidou et al., 2014; Saharinen et al., 2017; Schmittnaegel et al., 2017). Studies demonstrated that tumor angiogenesis is regulated by several factors and mechanisms and does not solely dependent on VEGF-A signaling (Bergers and Hanahan, 2008; Ferrara, 2016; Potente et al., 2011). Several studies have also shown the existence of tumor growth requiring no angiogenesis (Pàez-Ribes et al., 2009; Rubenstein et al., 2000).
Angiogenesis-independent tumor growth is also achieved through the action of immune cells such as macrophages and neutrophils (De Palma and Lewis, 2013; Gabrusiewicz et al., 2014; Liang and Ferrara, 2016). Coupling anti-angiogenesis specific therapy with strategies that disrupt any compensatory factors and signaling from cells such as macrophages and neutrophils offers better potential at inhibiting angiogenesis in tumors (Baer et al., 2016; Biswas and Mantovani, 2010; Lewis et al., 2016). Blockage or elimination of both macrophages and neutrophils that confer VEGF-A-independent angiogenesis to tumors has been shown to enhance tumor response to VEGF-A specific therapy (Rivera et al., 2015).
Kaneda et al. (2016) demonstrated that the inhibition of PI3K signaling, highly expressed in myeloid cells, successfully inhibited angiogenesis in cancer treatment. Caution must be taken, however, as several studies have shown that the inhibition of angiogenesis may enhance the ability of tumor-associated cells to promote tumor growth (Bergers and Hanahan, 2008; De Palma and Lewis, 2013; Ferrara and Adamis, 2016). Crawford et al. (2009) demonstrated that some tumors can overcome angiogenesis inhibition through upregulation of PDGF-C
Currently, Bevacizumab (anti-VEGF) is under clinical trials for the treatment of different cancers from colorectal cancer (NCT02885753), lung cancer (NCT03100955) to cholangiocarcinoma cancer (NCT03251443) in combination with 5-fluorouracil and panitumumab. Ramucirumab, also known as LY3009806, is under clinical trial in combination with paclitaxel for the treatment of gastric adenocarcinoma (NCT02898077). RAD001 is also under clinical trial for the treatment of renal cell carcinoma (NCT01206764).
CAFs as Delivery Vehicles of Therapeutic Agents
As described by Miao et al. (2017), CAFs can be used to deliver anti-tumor drugs or nanoparticles and cause cancer cell death via apoptosis. It has also been shown that fibroblasts within a tumor can scavenge and accumulate therapeutic drugs, resulting in increased drug concentrations within tumors (Hessmann et al., 2018). Senthebane et al. (2017) demonstrated that MSCs can be transformed into CAFs and thus are a major source of CAFs found in tumors. The authors and others demonstrated that although MSCs initially demonstrate anti-tumor behavior they are easily transformed into tumor-supporting cells (CAFs) when in contact with cancer cells (Dzobo et al., 2016b; Shi et al., 2017).
Due to their likelihood of being found within tumors, MSCs have been suggested as drug-delivery vehicles or can be manipulated to secrete anti-tumor factors (Shi et al., 2017). Amniotic MSCs have been suggested to deliver therapeutic agents to tumors (Bonomi et al., 2015). Several clinical trials on the use of MSCs in gene therapies for different cancers are underway (Abrate et al., 2014; Jobst et al., 2017; Niess et al., 2015). Overall, the use of MSC-based gene therapies requires further investigations as Senthebane et al. (2017) demonstrated. The authors' results show that MSCs can be pro-tumorigenic or anti-tumorigenic depending on tumor stage (Dzobo et al., 2016b; Senthebane et al., 2017). Other studies support this dual effect of MSCs (Clarke et al., 2015; Hong et al., 2014; Park et al., 2008).
Of late, CAFs are now being used as a stromal indicator of the disease stage during diagnosis and as an indicator of tumor response to treatment strategies. Tsujino et al. (2007) demonstrated that the number of myofibroblasts within TME can be used as a clinical biomarker of disease relapse in colorectal cancer. Surowiak et al. (2007) showed that the presence of CAFs within breast cancer tissue is a poor prognostic factor. Contrasting data on the prognostic value of CAFs in tumors may be due to CAFs' heterogeneity as well as the co-evolution of CAFs with tumor cells during tumor progression (Brechbuhl et al., 2017; Paulsson and Micke, 2014).
Outlook and Conclusion
To summarize, we provide the definitions of the key concepts in the present analysis in Box 1. Several lines of research are being initiated to evaluate the effectiveness of many therapeutic drugs on CAFs. Epigenetic drugs, including histone deacetylase inhibitors, are under investigation for their effect on several signaling pathways such as the JAK-STAT pathway in both cancer cells and CAFs (Dzobo, 2019). These epigenetic drugs are being evaluated on their potential to prevent CAFs' generation and increase in abundance within tumors (Younes et al., 2016). Another appealing strategy includes the use of normal fibroblasts in cancer treatment or the reversion of activated CAFs to normal fibroblasts (Kirsner et al., 2012).
Definitions of the Key Concepts in the Present Expert Review
Many questions remain to be answered regarding the origin of CAFs or their subsets, as this can influence CAF-directed therapeutic strategies adopted during cancer treatment. It is hoped that as new information becomes available, new markers and signaling pathways specific only to CAFs or their subsets can be identified.
The identification that MSCs, besides fibroblasts, can be a source of CAFs might explain CAFs' heterogeneity and could be the reason why several CAFs subsets are observed in tumors (Senthebane et al., 2017). If different cells contribute to the CAFs' subsets observed in tumors, which cell(s) of origin gives rise to pro-tumorigenic CAFs and which cell(s) gives rise to anti-tumorigenic CAFs? Based on data revealed by Senthebane et al. (2017), it appears that despite different cell(s) of origin, stromal cells may start as anti-tumorigenic but are transformed through several secreted factors to become pro-tumorigenic. Whether different cell types contribute to different subsets requires further investigation.
In addition, the identification of specific markers for different subsets can allow retrospective determination of cells of origin as well as the development of specific anti-tumorigenic therapies against these CAFs' subsets. Stromal cells can also co-evolve with tumor cells. Is tumor cell heterogeneity also partly driven by different CAF subsets? Epigenetic regulation of CAFs must also be investigated to allow the development of drugs targeting epigenetics of CAFs (Dzobo, 2019). Many clinical trials conducted for different candidate drugs have focused on targeting cancer cells and up to now no trial has been done on candidate drugs targeting CAFs.
One major challenge faced by scientists during the development of novel cancer therapies has been the absence of adequate cancer models that recapitulate the TMEs as seen in vivo to use in preclinical studies. This has prevented the proper understanding of tumor cell
New advances, including microfluidic technology, will allow the development of what is known as “human organs on chips.” With the ability to change parameters as required during tumor development as well as potential additions of several cells, including CAFs, these new cancer models will potentially reveal accurate information about tumor initiation and development. In addition, microfluidic technology as applied to tumor development will also allow collection of samples at specific times with ease.
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
The funding for this research was provided by the National Research Foundation (NRF) of South Africa (Grant no. 91457: RCA13101656402), the International Centre for Genetic Engineering and Biotechnology (ICGEB) (Grant no. 2015/0001), and the University of Cape Town.