
Abstract
The scientific basis of intracranial aneurysm (IA) formation, its rupture and further development of cerebral vasospasm is incompletely understood. Aberrant protein expression may drive structural alterations of vasculature found in IA. Deciphering the molecular mechanisms underlying these events will lead to identification of early detection biomarkers and in turn, improved treatment outcomes. To unravel differential protein expression in three clinical subgroups of IA patients: (1) unruptured aneurysm, (2) ruptured aneurysm without vasospasm, (3) ruptured aneurysm who developed vasospasm, we performed untargeted quantitative proteomic analysis of aneurysm tissue and serum samples from three subgroups of IA patients and control subjects. Candidate molecules were then validated in a larger cohort of patients using enzyme-linked immunosorbent assay. A total of 937 and 294 proteins were identified from aneurysm tissue and serum samples, respectively. Several proteins that are known to maintain structural integrity of vasculature were found to be dysregulated in the context of aneurysm. ORM1, a glycoprotein, was significantly upregulated in both tissue and serum samples of unruptured aneurysm patients. We employed a larger cohort of subjects (n = 26) and validated ORM1 as a potential biomarker for screening of unruptured aneurysms. Samples from ruptured aneurysms with vasospasm showed significant upregulation of MMP9, a protease, compared with ruptured aneurysms without vasospasm. We validated MMP9 as a potential biomarker for vasospasm in a larger cohort (n = 52). This study reports the first global proteomic analysis of the entire clinical spectrum of IA. Furthermore, this study suggests ORM1 and MMP9 as potential biomarkers for unruptured aneurysm and cerebral vasospasm, respectively.
Introduction
Intracranial aneurysm (IA) is a pathological dilation of intracranial artery that is characterized by focal deterioration of the vessel wall that involves loss of internal elastic lamina, disruption of media, and disintegration of adventitia and extracellular matrix of the artery (Nakajima et al., 2000; Rajesh et al., 2004). IA may be either familial or sporadic and is found to affect 3.2% of the general population (Vlak et al., 2011). Patients who harbor IA are usually not detected and are manifested only once the aneurysm ruptures, leading to intracranial bleed and elevated intracranial pressure (Zoerle et al., 2015). The consequences of rupture of an aneurysm are both on the cerebral and systemic physiology leading to multisystem changes (Chen et al., 2014).
Despite technological advances in the management for prevention of rebleeding, the blood present in the intracranial space itself acts as a trigger for various inflammatory pathways leading to delayed cerebral ischemia and worsens outcomes (Archavlis et al., 2013; Chaichana et al., 2010). Although the clinical spectrum of the disease has been established, pathophysiology of disease initiation and progression, that is, aneurysm formation, rupture, and subsequent development of cerebral vasospasm, remain largely unknown.
There are no molecular biomarkers that can be used for early detection of IA. Moreover, the patients at risk of aneurysmal rupture and those who subsequently develop cerebral vasospasm cannot be predicted. A few targets for disease management have shown promise but have not been able to reduce the disease burden significantly. It is intriguing that despite various scientific and technological advances in research and clinical management, it has not been possible to reduce disease burden that carries mortality of 30% and morbidity of 50% (Findlay et al., 2016).
Unraveling the molecular basis of different clinical subsets of IA may lead to the identification of potential biomarkers and therapeutic targets. Structural changes that are seen during aneurysm formation and rupture seem to be related to proteins as demonstrated in a few studies targeting single proteins (Connolly et al., 1997; Gaetani et al., 1997). Therefore, we undertook a global, untargeted proteomic study of entire disease spectrum of patients with IA using both aneurysm tissue and serum samples. We used a mass spectrometry (MS)-based quantitative proteomics approach to study protein-based molecular alterations in patients with IA. To our knowledge, this is the first study reporting whole proteome characterization of the entire disease spectrum in IA.
Materials and Methods
The study was designed to unravel the proteomic differences driving IA formation, rupture, and subsequent development of vasospasm. Consequently, four groups of patients were studied: (1) control group (C), (2) unruptured aneurysm (T1), (3) ruptured aneurysm without vasospasm (T2), and (4) ruptured aneurysm with vasospasm (T3). Subjects were recruited for this study from the departments of Neurosurgery, Neurology, and Forensic Medicine at the Post Graduate Institute of Medical Education and Research, Chandigarh. This study was approved by Institutional Ethics Committee PGIMER, Chandigarh (IEC-07/2015-268). All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Subjects were enrolled in the study only after obtaining an informed consent from the subject or their next of kin. Aneurysm tissue and blood samples were collected from the subjects of the four groups. For all the test groups, patients were diagnosed based on CT scan and angiographic examination. For controls, subjects with no evidence of IA on autopsy or angiographic examination were included for the study.
Aneurysm tissue samples
Aneurysmal wall tissue samples were collected from each test group during surgery. For controls, intracranial artery tissue was collected from the circle of Willis of subjects undergoing autopsy. Samples were collected within 4–6 h after death. Tissue samples were collected, washed with saline, and stored at −80°C until further use.
Serum samples
For test groups, blood samples were collected before surgery, before giving any anesthetic agent, or any treatment. For controls, blood samples were collected from subjects with negative angiography for any kind of cerebrovascular dysfunction. Serum was separated and stored at −80°C until further use.
Study design
Study was divided into two phases.
Discovery
In discovery phase, differentially regulated proteins were identified in aneurysm tissue and serum samples using untargeted proteomics. We collected a total of 20 tissue samples and 20 serum samples for discovery phase. Of them, five tissue/serum samples were collected in each test group (T1, T2, and T3) and control group (C).
Validation
In the validation phase, candidate biomarkers obtained from discovery phase were confirmed on a larger cohort of samples. For validation phase, a total of 78 serum samples were collected and processed in four groups, that is, C, T1, T2, and T3 (Table 1).
Demographic Data
Values are given as mean ± SD or number of patients.
F, female; M, male; SD, standard deviation.
Proteomic analysis
Schematic of the methodology is given in Figure 1. All chemicals were procured from Sigma Aldrich unless specified.
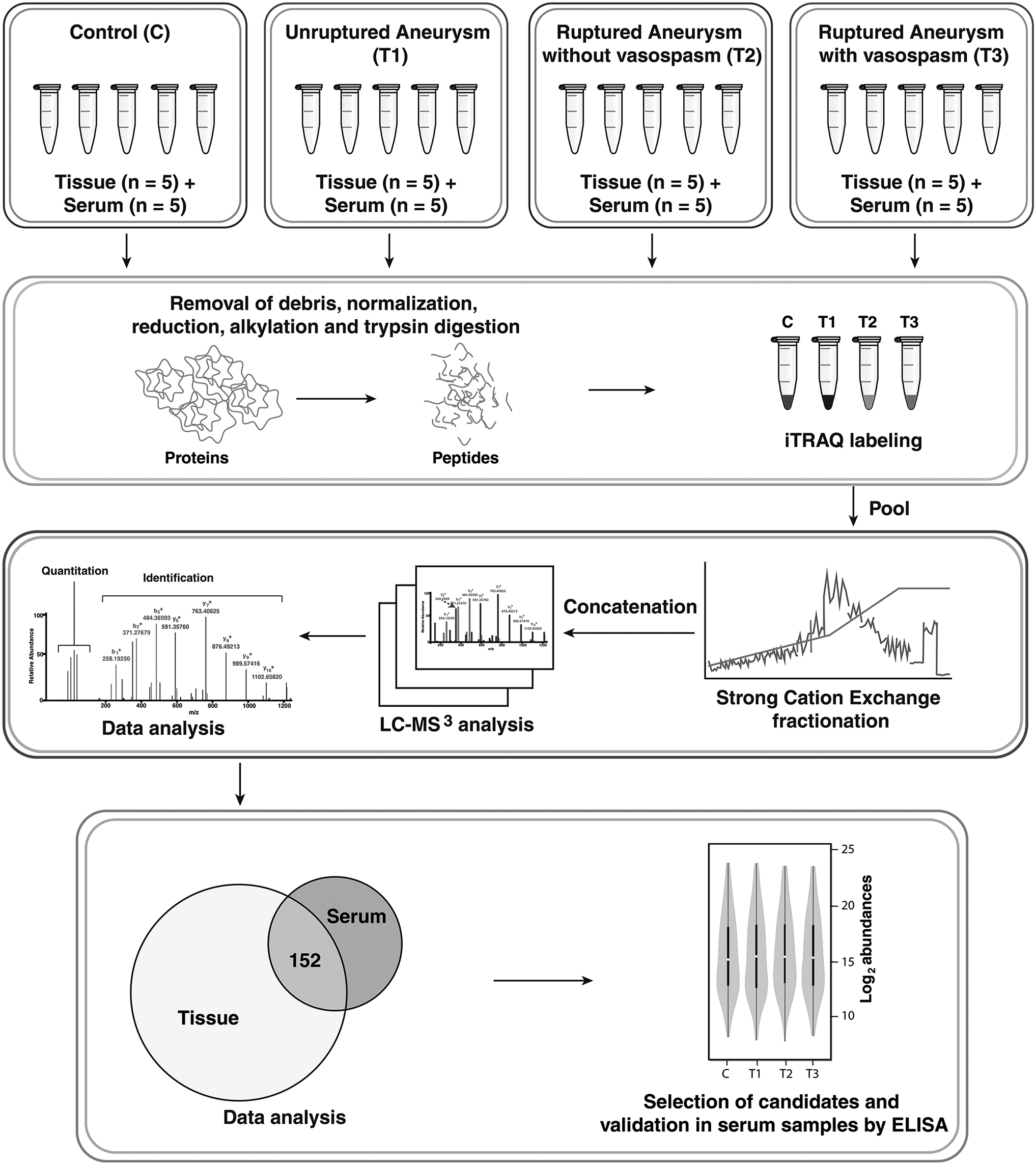
Study design and workflow.
Protein extraction
Equal amounts of five tissue samples from each group, that is, C, T1, T2, and T3 were pooled and disrupted in liquid nitrogen using a handheld disruptor. Subsequently, these were homogenized in lysis buffer (0.5% sodium dodecyl sulfate [SDS], 100 mM triethyl ammonium bicarbonate), centrifuged, and clear supernatant was separated. Protein estimation was carried out using bicinchoninic acid (BCA) assay (Pierce, Waltham, MA, USA).
For serum samples, total protein was measured by BCA assay in groups C, T1, T2, and T3. Equal amount of proteins from five individual samples were pooled in each group. Serum samples were then depleted of high abundant proteins using human multiple affinity removal system-14 column (Agilent Technologies, CA, USA) as per manufacturer's instructions.
Trypsin digestion
Equal amount of protein from each group was reduced using Tris (2-carboxyethyl) phosphine at 60°C for 1 h. Reduction was followed by alkylation using methyl-methanethiosulfonate at room temperature for 10 min. Trypsin (modified sequencing grade; Promega, Madison, WI, USA) was added (enzyme:protein = 1:20) to the samples and digestion was carried out at 37°C for 16 h. The reaction was quenched by acidifying the tryptic peptides with formic acid.
iTRAQ labeling and fractionation
4-plex iTRAQ (Sciex, Redwood City, CA, USA) was used to label the peptides as per manufacturer's instructions. Peptides obtained from group C were labeled with isobaric group 114, whereas peptides for groups T1, T2, and T3 were labeled with isobaric groups 115, 116, and 117, respectively. Labeled samples were pooled and subjected to strong cation exchange chromatography-based fractionation. A polystyrene-divinyl benzene copolymer modified with sulfonic acid Stage Tips (Empore Solid Phase Extraction Disk) were used as columns for fractionation. Sample digest containing iTRAQ-labeled pooled peptides was reconstituted in 70 μL of 1% TFA and fractionated into 6 fractions with in-house prepared columns. The fractions were dried, reconstituted in 0.1% formic acid, and desalted using C18 Stage Tips.
LC-MS3 analysis
iTRAQ-labeled fractions were analyzed on an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific, Bremen, Germany) interfaced with Proxeon Easy-nLC II system (Thermo Scientific). Each fraction was first loaded on a 2 cm long trap column packed with MagicC18AQ (Michrom Bioresources, Inc., Auburn, CA, USA). A linear gradient of 5–30% of solvent B (95% acetonitrile, 0.1% formic acid) at a flow rate of 300 nL/min was used on an analytical column (75 μm × 20 cm, Magic C18AQ) for separation of peptides. Total run time was set to 120 min. Each fraction was analyzed three times.
Mass spectrometer was operated on data-dependent acquisition mode. Scan range was set on 400–1600 m/z. Orbitrap mass analyzer was operated at a mass resolution of 120,000 at MS level. For MS/MS and MS/MS/MS levels, resolution was set to 30,000 and 60,000, respectively. Most intense precursor ions were selected and fragmented using higher energy collision dissociation.
Data analysis
The data acquired were searched against NCBI Human RefSeq81 protein database. The searches were performed using SEQUEST and MASCOT through Proteome Discoverer (version 2.1) software suite (Thermo Scientific).
The search parameters included trypsin as the proteolytic enzyme with one missed cleavage and oxidation of methionine as a dynamic modification. Static modifications were carbamidomethylation of cysteine and iTRAQ label at N-terminus of the peptide and lysine. Precursor and fragment ion mass tolerance were set at 10 ppm and 0.05 Da, respectively. False discovery rate (FDR) was calculated using a target-decoy strategy and 1% FDR was applied at the peptide level. Quantification was carried out using the reporter ion quantifier node for iTRAQ available in Proteome Discoverer. The MS raw data were deposited to the ProteomeXchange Consortium with the dataset identifier PXD013442.
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) kits were procured from Cloud-Clone Corporation (USA) and experiments were carried out as per manufacturer's instructions.
Statistical and bioinformatics analysis
For proteomics data, multiple t-tests with Holm–Sidak corrections were used for assessing the statistical significance between groups. Proteins that qualified fold change cutoff (FC ≥2) and statistically significant (p ≤ 0.05) were considered differentially regulated.
ELISA data were analyzed using a linear regression model. Unpaired t-test was used for assessing the statistical significance between groups. The diagnostic value of the candidate protein was further assessed using receiver operating characteristic curve analysis and area under the curve (AUC) calculations.
Heat maps were generated by Morpheus software (https://software.broadinstitute.org/morpheus) using hierarchical clustering method.
Results
In a quest to decipher the pathophysiology of aneurysm formation and identify potential biomarkers for screening and differential diagnosis of IA, we performed iTRAQ-based quantitative proteomic analysis of aneurysm wall tissue and serum samples of three groups of patients with IA and controls.
Demographic data
For discovery phase, we used 20 tissue and 20 serum samples in the 4 groups: C, T1, T2, and T3. For validating potential biomarkers, we included a larger cohort of 78 patients (13 control subjects, 13 patients of T1 group, and 26 patients each from T2 and T3 groups). Demographic details of the subjects recruited in the study are provided in Table 1.
Overview of proteomics data
In the past few years, quantitative proteomic analysis using isobaric mass tags has become the method of choice to compare proteomes of multiple physiological conditions. In this study, we used iTRAQ, one such robust isobaric tagging method for quantitative proteomics of IA. To improve the depth and breadth of the study, we used both tissue and serum samples of IA.
A total of 937 proteins were identified after tissue analysis. From serum samples, we identified 294 proteins, and 152 proteins were identified both from tissue and serum samples. Detailed information about the identified proteins in tissue and serum samples is provided in Supplementary Tables S1 and S2, respectively.
Principal component analysis of both tissue and serum data showed a clear separation between the test groups and controls (Fig. 2). We used unsupervised clustering to identify molecular signatures associated with the clinical subsets of this study (Fig. 3). We observed that the three clinical subsets were molecularly differentiated in both tissue (Fig. 3A) and serum (Fig. 3B) samples. Furthermore, several signature molecules were commonly found in tissue and serum and are described in the Results and Discussion Sections.

Principal component analysis.
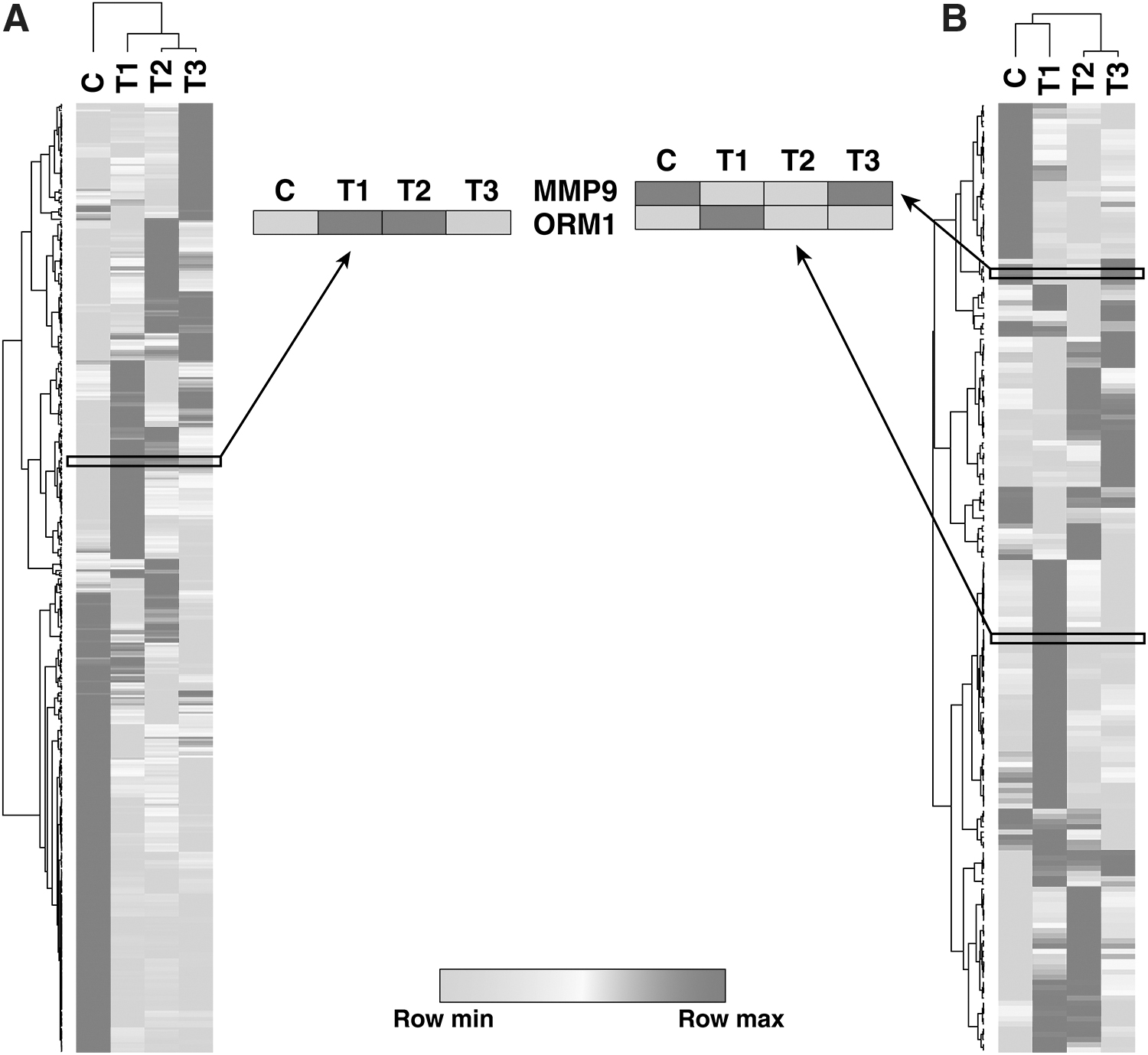
Heat map depicting molecular signatures associated with clinical subsets of patients with IA in
Differentially abundant proteins in the three groups of IA patients and their clinical potential
To identify the molecular events that led to the formation of IA, we analyzed the differentially regulated proteins of unruptured aneurysm patients in comparison with controls (T1/C). Several dysregulated proteins were commonly identified in both tissue and serum. Earlier studies have shown that inflammatory pathways and activation of the complement cascade are important factors during aneurysm formation (Taylor et al., 2015; Turkmani et al., 2015). Numerous proteins known to be involved in inflammatory processes and complement cascades were found to be overexpressed in this study. Such proteins include α-2-macroglobulin, complement factor H, complement C3, complement C1q, complement C5, S100A4, and S100A9.
Thrombospondins are involved in maintaining the structure of vasculature by mediating cell-to-cell and cell-to-matrix interactions (Resovi et al., 2014). Thrombospondin 1 (THBS1) is known to contribute to the development of abdominal aortic aneurysm (AAA) by causing vascular inflammation through regulation of migration and adhesion of mononuclear cells (Liu et al., 2015). Thrombospondin 4 (THBS4) is involved in local signaling in nervous system and contributes to spinal sensitization and neuropathic pain state (Kim et al., 2012). Role of THBS4 in inducing AAA has been recently demonstrated (Palao et al., 2016). In our study, THBS1 and THBS4 were overexpressed in tissue and serum, respectively. Thus, thrombospondins may play a role in the formation of IA.
A significantly high number of patients with IA suffer from aneurysmal rupture every year. Comparing protein expression profiles of ruptured aneurysms with unruptured aneurysms is likely to provide insights into molecular mechanisms underlying IA rupture. Chronic inflammation has been associated to aneurysm rupture in earlier studies (Hashimoto et al., 2006; Hudson et al., 2013). We observed overexpression of proteins related to inflammatory pathways in our study. C-reactive protein was found to be upregulated in our study, which has been associated with an increase in size of AAA and its rupture (Turner et al., 2015; Vainas et al., 2003). Similarly, there was overexpression of SAA1 protein that could lead to aneurysmal rupture (Webb et al., 2015).
Development of vasospasm is seen to be detrimental to favorable prognosis postaneurysmal rupture (Findlay et al., 2016). Proteins that play a role in the formation of vasospasm may be used as potential prognostic biomarkers. To identify such molecules, we compared protein expression profiles of ruptured aneurysm with vasospasm and ruptured aneurysm without vasospasm (T3/T2). To our knowledge, this is the first proteomic study of these two clinical subsets. Superoxide has been reported to be associated with vasospasm (Mori et al., 2001). We observed overexpression of Cytochrome b-245 (CYBA). CYBA is known to associate with NOX3 to form a functional NADPH oxidase, which leads to generation of superoxide (Ueno et al., 2005).
We also observed overexpression of proteins related to oxidative stress, such as myeloperoxidase, which has been associated with acute ischemic stroke (Cojocaru et al., 2010). Monoamine oxidase-A was also found to be overexpressed. This protein has been demonstrated to oxidize biogenic amines such as norepinephrine and 5-hydroxytryptamine causing oxidative stress. In serum data, we observed elevated expression of peroxiredoxin-2. This protein is related to oxidative stress and was found markedly elevated in the brain extracellular fluid in patients with acute ischemic stroke (Dayon et al., 2011).
ELISA-based validation
ORM1
ORM1 is a glycosylated protein that binds to, and transports basic and neutral molecules (Fournier et al., 2000). ORM1 is an acute phase anti-inflammatory protein that also plays a role in injury-induced angiogenesis (Ligresti et al., 2012). In this study, ORM1 was found to be 22-fold overexpressed in tissue and 9-fold upregulated in serum of patients with unruptured aneurysm and was selected as a candidate molecule for validation.
ELISA-based validation showed that levels of ORM1 were significantly higher in group T1 compared with C. The AUC was found to be 0.78 with sensitivity and specificity of 76.92% and 84.62%, respectively (Fig. 4). Thus, ORM1 may be a potential biomarker for population screening of unruptured IA.
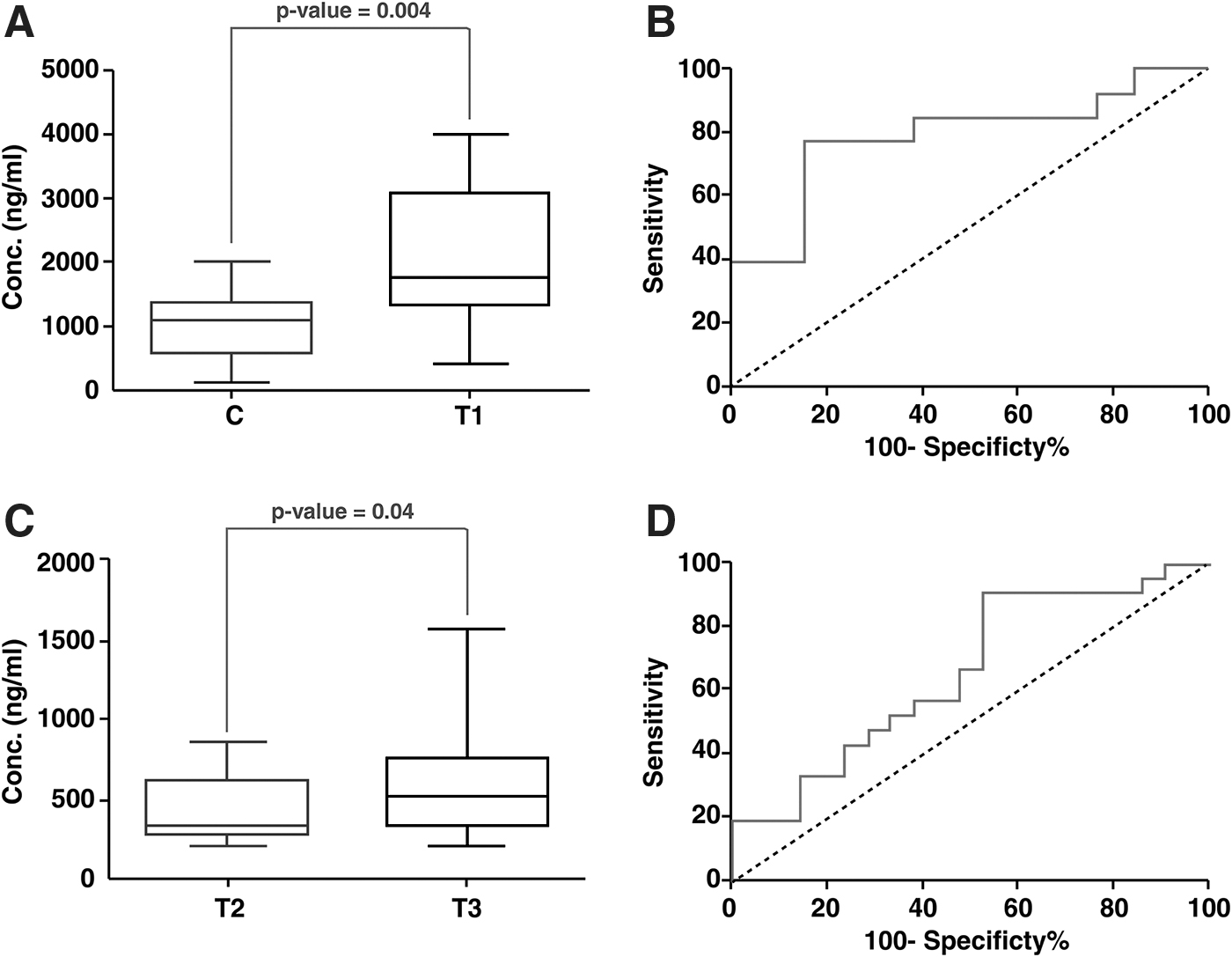
Estimation of ORM1 and MMP9 expression by ELISA. Box plots depicting the difference in the expression level of
Matrix metalloproteinase 9
Matrix metalloproteinases (MMPs) are a family of extracellular and membrane-bound proteases that degrade or proteolytically modify the extracellular matrix through interactions with collagenases, laminins, and proteoglycans (Löffek et al., 2011). MMP9 was found to be 13.4-fold upregulated in serum samples of patients in group T3 compared with T2 and was selected as a candidate molecule for validation.
ELISA of serum samples showed that levels of MMP9 were significantly higher in T3 as compared with T2. The AUC was found to be 0.64 with sensitivity and specificity of 68.18% and 50%, respectively (Fig. 4).
Discussion
MS-based quantitative proteomics has emerged as the method of choice to identify molecular signatures of disease. A few studies have carried out MS-based analysis of IA (Jiang et al., 2018; Wang et al., 2015, 2016; Xu et al., 2015). To our knowledge, there is no study depicting the differential proteomic profile of the three subgroups of patients.
To delineate the pathophysiology of aneurysm formation, rupture, and progression to vasospasm, MS-based analysis was performed. We analyzed aneurysm tissue and serum samples of patients with IA using quantitative proteomics. We chose to analyze both tissue and serum to look for the reflection of dysregulated tissue proteins (microenvironment of disease) in serum samples (macroenvironment of disease). This would help us in identifying biomarkers in serum samples that is an easily accessible biofluid. This study has resulted in the identification of numerous clinically interesting proteins that may have significant contribution to the genesis and progression of disease.
Pathophysiology of aneurysm formation
Despite being a dreadful complication of brain vasculature, the pathogenesis of IA formation remains poorly understood. Although the presence of IA is significant in the general population (Vlak et al., 2011), we are still at loss in detecting such patients before aneurysm rupture. Studying the biology of aneurysm formation may decipher molecules that can serve as biomarkers for population screening. In this study, proteins such as ORM1, SERPINA1, and SAA2–SAA4, were overexpressed in both the tissue and serum. These have a role in the inflammatory pathways and inflammation has been demonstrated as an important pathological factor for formation of IA (Chalouhi et al., 2012; Hosaka and Hoh, 2014).
Proteins that are crucial for organizing cytoskeleton and its maintenance were found to be downregulated, for example, myosin-11, filamin-A, aortic smooth muscle, smoothelin, α-actinin-4, tropomyosin1, tubulin, LAMB2, and ACTN1. They play a significant role in cell-to-cell communication, interaction, and regulation of movement. Their altered expression may explain the thinning of muscular layer of the intracranial vessel wall at the site of aneurysm formation. Cadherin 2 was also downregulated in our study. Cadherins protect the cells from mechanical shear and stress and its deficiency may result in the loss of the protective ability against wall shear stress (WSS). WSS has been reported to play a role in the formation of IA (Chatziprodromou et al., 2007).
ORM1 is a potential screening biomarker for unruptured IA
Quantitative proteomic profiling of IA tissue and serum samples in patients with unruptured aneurysm and controls identified ORM1 as a potential biomarker for aneurysm formation. ELISA-based validation also confirmed its upregulation in serum of patients with unruptured aneurysm compared with controls.
ORM1 plays an important role in injury-induced angiogenesis (Ligresti et al., 2012). At the site of vascular injury, endothelial cells on the luminal side are persuaded by tumor necrosis factor-α (TNF-α) (secreted from macrophages) to secrete ORM1. ORM1 inhibits the TNF-α at the site of injury acting as an anti-inflammatory protein. At the same time, it induces angiogenesis. A well-known pathophysiological consequence of injury-induced angiogenesis is that it increases permeability of intercellular endothelial cell junctions leading to edema and extensive injury to the surrounding tissue (Weis and Cheresh, 2005). Thus, ORM1 may play a role in aneurysm formation by inducing angiogenesis at the site of intimal injury that may progressively damage the vessel wall tissue (Fig. 5).
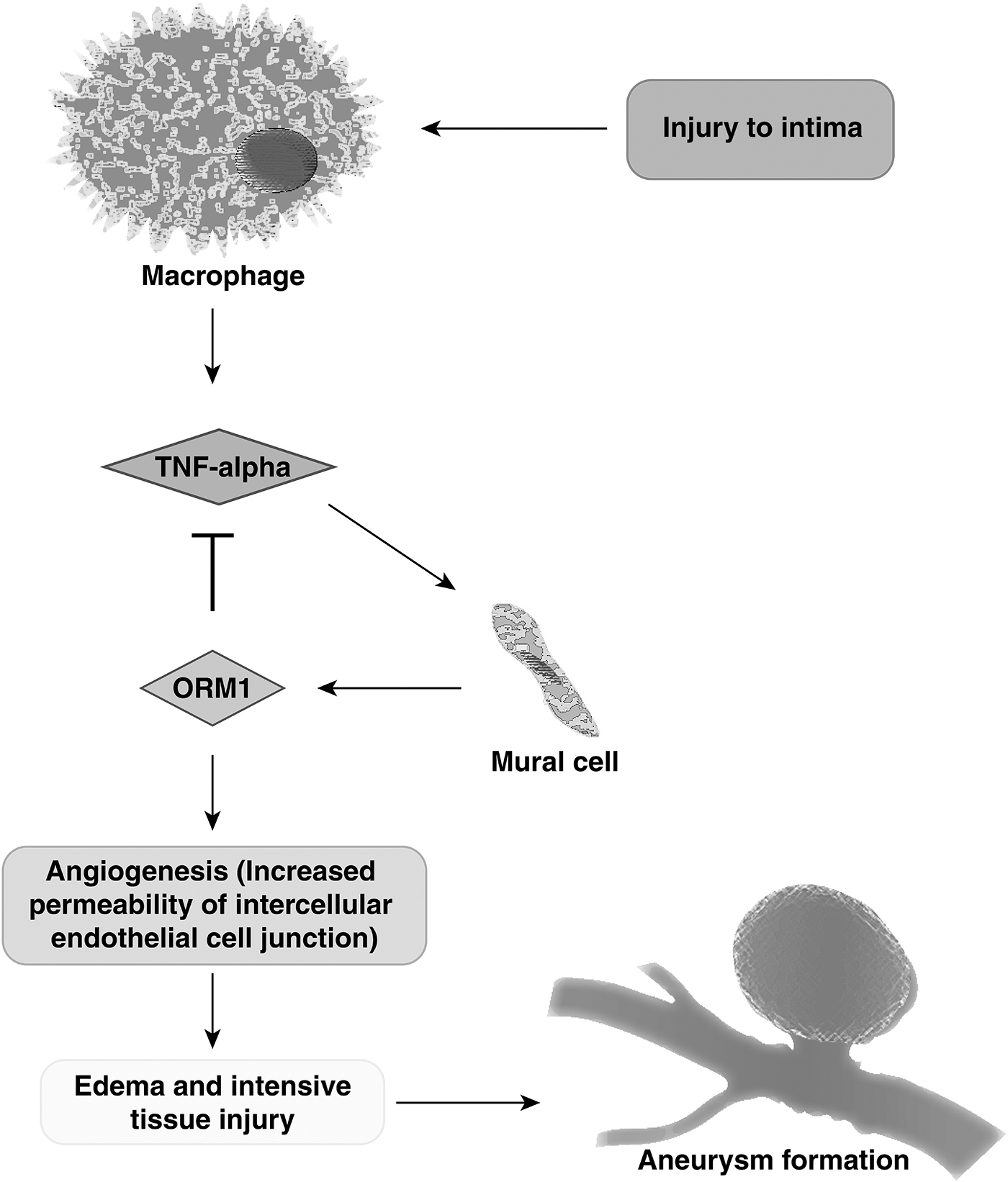
Potential mechanism of aneurysm formation through ORM1.
Molecular mechanisms underlying aneurysmal rupture
There has been lot of enthusiasm in trying to understand how aneurysms formed in the cerebral vasculature eventually progress to rupture. It is intriguing why only few of the aneurysms rupture and most of them do not. To understand the biology of aneurysmal rupture, we studied differential expression of proteins in tissue and serum samples of patients with ruptured aneurysm and compared it with those with unruptured aneurysm.
We observed overexpression of Cathepsin B (CTSB), a lysosomal cysteine protease. It is possible that an increased activity of CTSB may contribute to aneurysm progression through caspase-induced apoptosis of vascular smooth muscle cells (VSMCs), as supported by previous reports (Aoki et al., 2008; Guo et al., 2016). Overexpression of fibrinogens (FGA, FGB, and FGG) found in this study is in good agreement with a previous report that demonstrated that an increased level of fibrinogens is a marker for aneurysm progression in AAA (Al-Barjas et al., 2006).
Proteomic aberrations leading to vasospasm
On comparing the protein expression profiles of patients with ruptured aneurysm with and without vasospasm (T3/T2), we observed overexpression of azurocidin (AZU1). AZU1 is expressed in endothelial cells favoring contraction and increased endothelial permeability (Gautam et al., 2001). Narrowing of blood vessels that lead to cerebral vasospasm is characterized by prolonged abnormal contraction of VSMCs (Chaichana et al., 2010; Findlay et al., 1991; Ladner et al., 2013). Thus, an increased expression of AZU1 points to the fact that contraction of blood vessel may lead to cerebral vasospasm.
S100A8 and S100A9 are calcium-binding proteins that are known to play an important role in the regulation of inflammatory processes and immune response. Overexpression of these proteins also indicates the importance of inflammation in the development of vasospasm. Proteins related to cell adhesion and remodeling of the actin cytoskeleton such as integrin family proteins (ITGA2B and ITGB3), neutrophil defensin 1, myeloblastin, and lactotransferrin were found to be upregulated in patients with vasospasm. Blocking of integrins by their receptor antagonist GRGDSP has been shown to prevent vasospasm in earlier studies (Pradilla et al., 2004), which is correlated well with our finding.
MMP9 is a potential biomarker for vasospasm
Quantitative proteomic profiling of serum enabled the identification of MMP9 as a potential prognostic biomarker for vasospasm formation and was validated on a larger cohort of patients using ELISA. MMP9 has been implicated in the pathophysiology of blood–brain barrier (BBB) disruption and cerebral edema (Turner and Sharp, 2016). The mechanism is complex, but on disruption of BBB, there is leakage of inflammatory mediators, which leads to arterial deregulation owing to leukocyte migration. This blockage in the artery may lead to its narrowing, resulting in vasospasm. Furthermore, minocycline, an inhibitor of MMP9, has been shown to decrease vasospasm (Vellimana et al., 2017). Overall, it can be concluded that MMP9 has the potential to be used as a biomarker and a therapeutic target.
Limitations
There are some limitations to this study. We had a small sample size in discovery phase. This was owing to the difficulty in obtaining tissue samples of aneurysm wall. However, validation was performed in a larger cohort based on sample size calculations. ORM1 and MMP9, the potential biomarkers identified, need to be tested in a larger, geographically distinct cohort of patients to determine their diagnostic potential.
Conclusions
This study provides the first whole proteome characterization of the entire spectra of IA using both tissue and serum samples. Multiple proteins that may have a role in the pathogenesis of aneurysm formation, rupture, and subsequent development of cerebral vasospasm have been elucidated. Findings from this study could lead to better diagnostic capabilities and therapeutic interventions.
Footnotes
Acknowledgments
The authors thank the patients and family members for their participation in the study. The authors would like to express their greatest gratitude to Late Dr. V.K. Grover for his invaluable guidance and support for conducting the study.
Author Disclosure Statement
The authors declare they have no competing financial interests.
Funding Information
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.