
Abstract
Carbonic anhydrase II (CAII) is one of the zinc metalloenzymes that catalyze the reversible hydration of carbon dioxide, leading to the formation of bicarbonate and proton. CAII plays a significant role in health and disease. For example, CAII helps to maintain eye pressure while regulating the pH of the tumor microenvironment, and by extension, contributing to cancer progression. Owing to its remarkable role in cancer, visual health, and other human diseases, CAII can serve as an attractive therapeutic target. We report an original study based on high-throughput virtual screening of natural compounds from the ZINC database in search of potential inhibitors of CAII. We selected the hits based on the physicochemical, absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties, pan-assay interference compound (PAINS) patterns, and interaction analysis. Importantly, two natural compounds were identified, ZINC08918123 and ZINC00952700, bearing considerable affinity and specific interactions to the residues of the CAII-binding pocket with well-organized conformational fitting compatibility. We investigated the conformational dynamics of CAII in complex with the identified compounds through molecular dynamics simulation, which revealed the formation of a stable complex preserved throughout the 100 ns trajectories. The stability of the protein/ligand complexes is maintained by significant numbers of noncovalent interactions throughout the simulations. In conclusion, natural compounds identified in the present study specifically and computer-assisted drug design broadly offer a reliable resource and strategy to discover potential promising therapeutic inhibitors of CAII to cure various cancers and glaucoma after further experimental validation and clinical studies.
Introduction
The pH of the tumor microenvironment plays a vital role in thriving the tumors (Estrella et al., 2013). Carbonic anhydrases (CAs) are regulators of the pH of the tumor microenvironment by adjusting the bicarbonate and proton concentration necessary for cell survival and growth of the tumor (Mboge et al., 2018). Carbonic anhydrase II (CAII) is a zinc metalloenzyme that catalyzes the conversion of carbon dioxide to carbonic acid (Hassan et al., 2013). This enzyme reversibly hydrates carbon dioxide to bicarbonate and protons. Thus, it is involved in regulating pH and maintains CO2 homeostasis and osmotic balance (Aditama et al., 2015). CAII mRNA expression is found to be upregulated in various cancers, such as breast, bladder, thyroid, liver, lung, pancreas, prostate, renal cell carcinomas, head, neck, etc. (Cerami et al., 2012).
CAII gene is present on chromosome 8 of humans, which translates into amino acid residues comprising of 7 α-helices (αA-αG) and 5 β-sheets (βA-βE) (Boriack-Sjodin et al., 1998; Dar et al., 2017). The catalytic active site has a 15 Å deep conical cleft, where the zinc ion is present, tetrahedrally coordinated to N atoms of three histidine residues (metal-binding sites His94, His96, and His119) (Saito et al., 2004). The active site (proton donor/acceptor) of CAII is His64. The active site of CAII is amphiphilic, featuring both hydrophilic and hydrophobic residues. His64, Asn67, Thr200, Asn69, Gln92, Thr199, Tyr70, and Val62, make the hydrophilic part of the cavity, whereas the hydrophobic part consists of Ala121, Phe131, Val207, Phe91, Lue131, Asp138, Phe146, Val109, Pro201, and Pro202 (Xue et al., 1993).
CAII is expressed in many organs, which is highly crucial for the normal functioning of a human body, including the brain, liver, stomach, kidney, etc. (Hassan et al., 2013). High expression of CAII has been found in the appendix, cerebral cortex, duodenum, colon, gall bladder, hippocampus in the brain, kidney, liver, rectum, seminal vesicle, salivary gland, and stomach. In contrast, a moderate expression of CAII is found in bone marrow, breast, cerebellum, lung, and pancreas (Dar et al., 2017). An upregulation in CAII level was observed in atopic dermatitis by polyinosinic:polycytidylic acid, a toll-like receptor 3 (TLR-3) protagonist through Th2 cytokines.
This study suggested that CAII is involved in pathways induced by Th2 and TLR-3 in inflammatory skin conditions (Kamsteeg et al., 2007). CAII is responsible for maintaining the inner eye pressure, and its failure is associated with glaucoma (Ghorai et al., 2020). Owing to its essential role in cancer progression and eye pressure maintenance, CAII represents a well-established therapeutic target for various complex diseases, including glaucoma and cancer (Ghorai et al., 2020; Mboge et al., 2018; Noor et al., 2018; Scozzafava and Supuran, 2014).
Many compounds, including sulfonamide derivatives, for example, acetazolamide, methazolamide, brinzolamide, ethoxzolamide, dichlorophenamide, dansyl amide, etc. have shown potent inhibitory effects on CAII and CAIX (Amresh et al., 2013; Ghorai et al., 2020; Kumari et al., 2016; Mboge et al., 2018; Noor et al., 2018; Queen et al., 2018; Scozzafava and Supuran, 2014). Despite their clinical accomplishment, resistance due to mutations and side effects is still a limitation of the available drugs (De Simone et al., 2005; LIANG et al., 1993). Their less effectivity and high toxicity have shifted the interest of scientists to explore other substitutes like natural compounds.
Naturally derived compounds are safe, economical, and compatible with humans compared with conventional synthetic drugs and thus encouraged to be used in anticancer drug development. To overcome resistance and to avoid side effects, novel inhibitors with higher potency and low toxicity are required (Aneja et al., 2020; Mushtaque et al., 2021; Shamsi et al., 2020; Supuran, 2012; Supuran et al., 2003). Structure-based drug discovery methodologies have proven their potential in the quick discovery of lead-like compounds from scratch (Ali et al., 2019; Ansari et al., 2018; Das Mahapatra et al., 2020; Idrees et al., 2018; Kumari et al., 2016). Virtual high-throughput screening (vHTS) approaches utilizing molecular docking, and molecular dynamics (MD) simulation have great potential in studying protein-ligand interactions and their stability (Naqvi et al., 2018).
The combination of molecular docking and MD simulation flexibly treats the protein-ligand complex and can examine the impact of water molecules to mimic the natural environment (Mohammad et al., 2020c). In this study, we aim to identify potential inhibitors of CAII using a computer-assisted drug discovery approach while utilizing molecular docking and MD simulations. The working flowchart of the applied vHTS process in the present study is illustrated in Figure 1.

Computational approach adopted to discover high-affinity inhibitors of CAII. CAII, carbonic anhydrase II; MD, molecular dynamics.
Materials and Methods
Computational tools and web resources
The virtual screening and MD simulation were performed on the HP Z420 workstation. Online resources like Protein Data Bank (Rose et al., 2012), ZINC database (Irwin and Shoichet, 2005), SwissADME (Daina et al., 2017), and the pkCSM webserver (Pires et al., 2015) were used for retrieving structural data and filtering compounds. A library of natural products containing five subsets of ∼90,000 virtual compounds was retrieved from the ZINC repository. SwissADME webserver was used for applying Lipinski's rule of five and pan-assay interference compounds (PAINS) filter. InstaDock (Mohammad et al., 2020a) was used for molecular docking-based vHTS.
InstaDock is an all-in-one GUI program that uses AutoDock Vina-based QuickVina-W software for docking calculation and provides a set of precalculated parameters, including pKi and ligand efficiency. Visualization of protein and ligand interactions was done by PyMOL (DeLano, 2002) and Discovery Studio Visualizer (Biovia, 2017). The all-atom MD simulation for 100 ns was performed for each system using the GROMACS suite (Van Der Spoel et al., 2005).
Physicochemical properties of the compounds
The ZINC database containing ∼90,000 natural compounds was converted into the SMILES formula using Discovery Studio Visualizer to calculate physicochemical properties. This library includes many compounds with destructive patterns, which might result in the selection of false positives. To avoid those compounds, we have screened the library based on their physicochemical properties. In this study, we have applied Lipinski's rule of five for filtering out the compounds. The physicochemical and PAINS properties of the compounds were calculated through the SwissADME server, and carcinogenicity was predicted using CarcinoPRED-EL. The library was also filtered out for those compounds containing PAINS patterns. This step helps us to get safer molecules with high drug likeliness. We also evaluated the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of the compounds while using the pkCSM webserver.
Structure-based virtual screening
Molecular recognition is the basis of drug activity (Dukhovich, 2004). The pharmacological effect of a ligand molecule is shown by its likeliness to bind to the target receptor and inhibit/activate its activity by forming a stable complex (Stocks, 2013). The process of forming a complex between ligand and receptor is a complex equilibrium process, where a small molecule binds to the protein occupying the binding site (Babine and Bender, 1997). Starting with the three-dimensional structure of CAII and available ligands, we have performed their docking and scoring that suggests us some hits, which show appreciable binding affinities. The structural coordinates of human CAII were downloaded from the PDB (PDB ID: 5NY6).
The receptor was prepared by deleting native water molecules and cocrystallized ligands from the original coordinates before the docking process. The grid center was identified using InstaDock, making the search space big enough for the ligands to rotate smoothly and find their most favorable binding sites. The grid box was defined in a blind search space manner with a spacing of 1 Å and size of dimensions 60 × 57 × 68, centralized at −9.69, −1.74, and 16.05, pointing at x, y, and z-direction, respectively.
The filtered library of natural compounds was then subjected to a molecular docking study with CAII. All compounds were processed in InstaDock before docking and calculating their binding affinities. All possible docking orientations of each ligand were searched toward CAII while utilizing the Vina-default algorithm (Trott and Olson, 2010). The binding affinity for each compound was calculated and ranked through the Vina scoring function embedded in InstaDock. After searching and scoring, the initial hits were identified based on their docking score, that is, binding affinity. The selected hits were further explored for interaction analysis while utilizing PyMOL and Discovery Studio Visualizer. The final hits were chosen on the basis of their specific interactions toward the CAII-binding site.
Prediction of activity spectra for substances analysis: biological activity predictions
The prediction of activity spectra for substances (PASS) analysis was carried out using the PASS webserver, which predicts thousands of pharmacological effects and biochemical mechanisms based on the molecular formula (Parasuraman, 2011). We have predicted the potential biological and pharmacological properties of the compounds using the PASS webserver. This server evaluates the biological potential of a compound based on its structure/activity relationship. It compares the input structure with an inbuilt training set with varying types of biological activities. The prediction gives a summarized list of probable biological activities on the ratio of “probability of being active (pa)” and “probability of being inactive (pi).” A higher pa value signifies the higher probability of a biological property for a compound being examined.
MD simulations
The selected binding poses of the identified compounds in complex with CAII were used as initial coordinates for MD simulation studies. A set of three simulations was carried for 100 ns each on free-CAII and CAII with two of the identified compounds, that is, ZINC08918123 and ZINC00952700. The topology parameters for both the ligands were generated from the PRODRG server. All simulations were carried out at 300 K temperature using the GROMOS 54A7 force-field embedded in the GROMACS-2020 beta-2.
First, all three systems were soaked in a cubic periodic box of the TIP3P water model with a 10 Å distance to the edges. Next, all three systems were electro neutralized by adding an appropriate number of counter ions [sodium (Na+) and chlorine (Cl−)] followed by an energy minimization process for 1500 steps of the steepest descent methods. Next, the equilibration for 1000 ps was carried out in two steps, that is, NVT [moles (N), volume (V), and temperature (T)] and NPT [moles (N), pressure (P), and temperature (T)]. Finally, the resultant systems from the NPT equilibration were subjected to a final MD production for 100 ns. The visualization and graph preparation were done while utilizing the VMD and QtGrace tools (Turner, 2009).
Principal component and free energy landscape analysis
Principal component analysis (PCA) has been a valuable tool to explore the principal motions of a protein. It has been widely utilized in exploring the folding dynamics of protein under stress conditions. We have performed PCA on alpha-carbon (Cα) atoms of CAII, CAII-ZINC08918123 and CAII-ZINC00952700 to explore their conformational sampling, atomic motions, and stability. The MD trajectory of all three systems was also utilized for free energy landscape (FEL) analysis by diagonalizing the eigenvectors (EVs) for the covariance matrix (Altis et al., 2008; Amir et al., 2019; Mohammad et al., 2019). The following formula was utilized in calculating the covariance matrix:
where xi/xj represents the ith/jth atom coordinates, and < - > is the ensemble average.
The FEL plots of CAII, CAII-ZINC08918123, and CAII-ZINC00952700 systems were generated through the conformational sampling method by exploring the structural variability and the native state (Altis et al., 2008). The FEL plots were analyzed to evaluate the conformational stability of CAII-apo, CAII-ZINC08918123, and CAII-ZINC00952700 complexes. The FEL plots were produced while utilizing the following formula:
KB signifies the Boltzmann constant, T is the simulation temperature, and P(X) signifies the PCs' probability distribution.
Results and Discussion
Physicochemical properties of the compounds
The parent library from the ZINC database contains ∼90,000 natural compounds, many with destructive patterns, resulting in the selection of false positives. Hereafter, calculating the physicochemical properties of all the natural compounds, we have applied the filter of Lipinski's rule of five. We have selected the compounds bearing ≤5 H-bond donors, no >10 H-bond acceptors, a molecular mass no >500 Da, and the logP value not greater than 5. Also, keeping in mind a drug's likeliness to enter a body and get absorbed, easily distributed, enters metabolism, and gets eliminated, we applied ADME filter cutting down more unlikable compounds. Finally, before the receptor-based virtual screening, CarcinoPred-EL was applied to check the carcinogenicity of the compounds. Finally, after applying these initial filters, a library of 32,902 compounds was utilized for further evaluation.
Binding affinity with CAII
The filtered library of 32,902 compounds was subjected to receptor-based virtual screening. All the compounds were docked to CAII one by one to estimate their binding affinities and possible interactions. Among the compounds docked, a set shows an appreciable binding affinity toward CAII (ΔG ≤ 13.6 kcal/mol). We extracted the top 10 compounds showing the affinity score in the range of −13.6 to −14.9 kcal/mol, which is higher than the affinity of the known cocrystallized CAII inhibitor ZINC00638494 [4-chloranyl-∼{N}-[(1∼{R})-1-(2-hydroxyphenyl)ethyl]-3-sulfamoyl-benzamide] (Table 1). ZINC08918123 showed the highest binding affinity of −14.9 kcal/mol. As filtered from the initial screening, all compounds follow Lipinski's rule and show drug likeliness. The chemical structures of the selected compounds, along with their SMILES strings, are given in Supplementary Table S1.
Binding Affinities and Physicochemical Properties of Top 10 Hits Along with the Crystallized Carbonic Anhydrase II Inhibitor (ZINC00638494)
Interaction study
While choosing the final hits, considering the protein/ligand complex's interacting residues is one of the crucial steps to avoid unspecific binding. Most of the cocrystallized inhibitors of CAII found to interact with His64, Thr199, and Thr200 and make polar interactions. The interaction analysis of the selected ten compounds was carried out by Discovery Studio Visualizer. The output files of selected compounds generated a total of 90 docked conformers. Finally, based on the specific interactions toward CAII, two compounds ZINC08918123 and ZINC00952700, were identified. Both compounds share common interactions with the cocrystallized inhibitor ZINC00638494 (PDB ID: 9EB) and fitted well into the binding pocket of CAII with a good complementarity (Fig. 2).
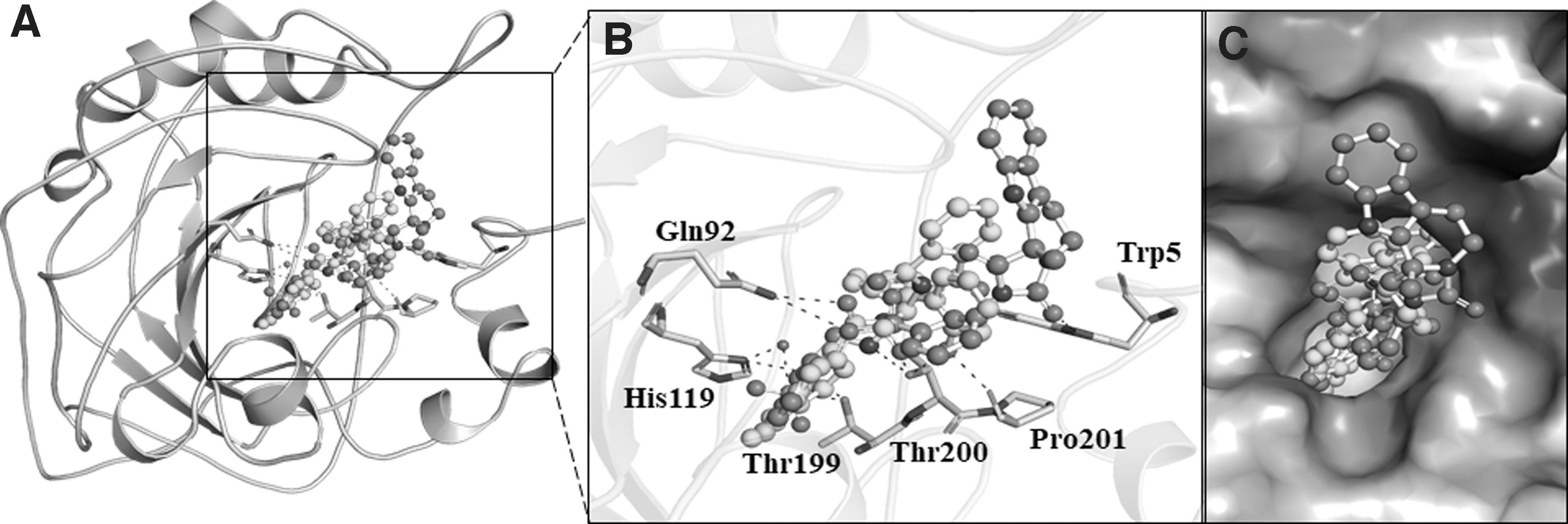
Binding pattern of the selected compounds with CAII.
Both compounds docked in the CAII-binding pocket were further explored for detailed interactions and their types. It was found that compound ZINC08918123 interacts with many close interactions, including a hydrogen bond (H-bond) with Thr200 (Fig. 3A). At the same time, compound ZINC00952700 was found to interact with three H-bonds with Asn67, Gln92, and Thr199 (Fig. 3B). Compound ZINC00952700 shares more common interactions with the cocrystallized inhibitor than ZINC08918123 (Fig. 3C). Both compounds, along with the cocrystallized inhibitor found to interact with the active site residue, His64, which in turn might inhibit the functional activity of CAII (Fig. 3). In most of the cases, rings are forming hydrophobic interactions to the residues of CAII in close contact.
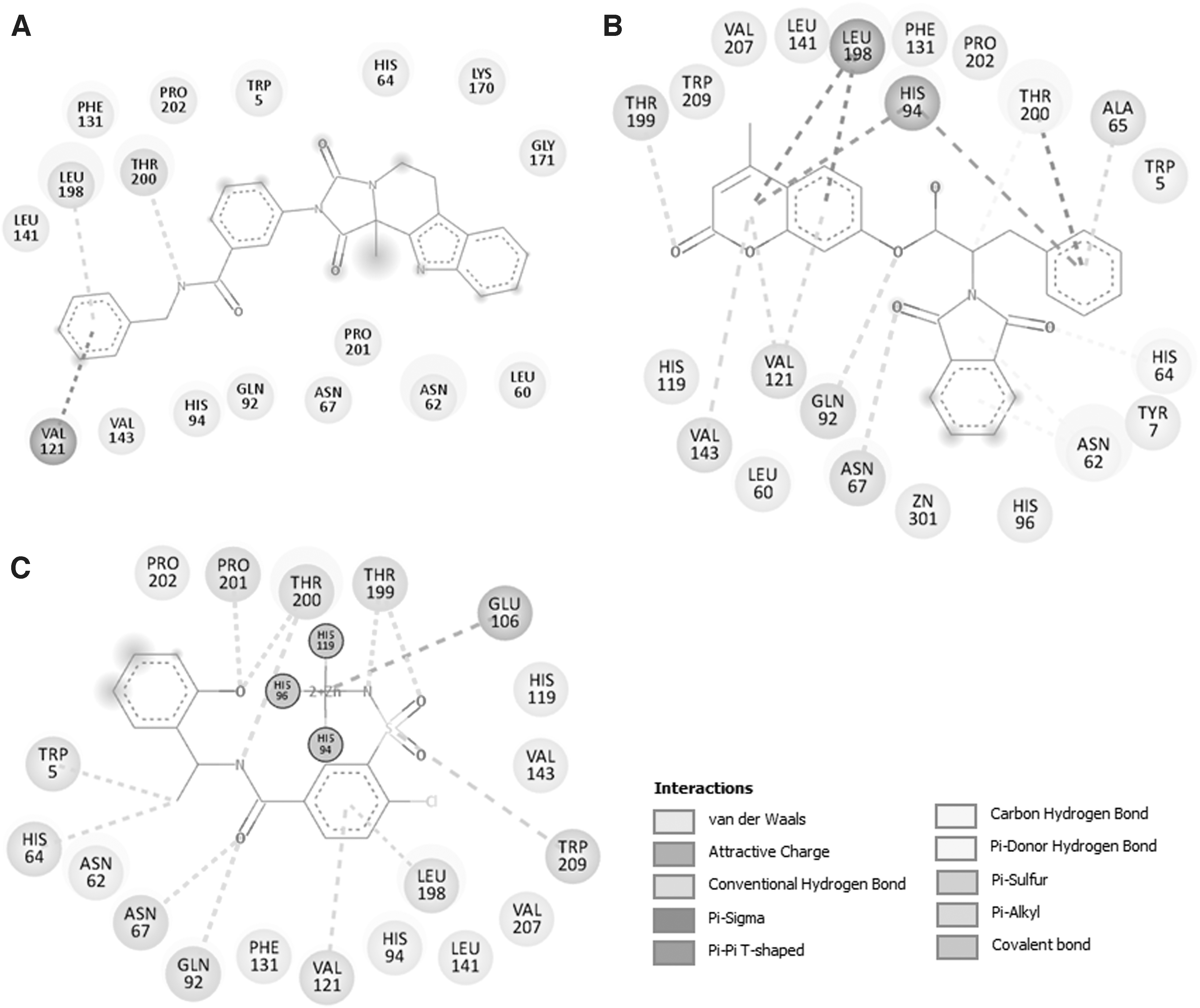
2D interaction plots of CAII residues with the finally selected compounds
However, C = O and N form hydrogen-bonded interactions to the Thr200, Thr199, Gln92, Asn67, His64, Trp209, and Pro201. Benzene sulfonamide inhibitor binds to CAII with sulfonamide group coordinated to the zinc ion in a tetrahedral geometry. In fact, the same group forms hydrogen-bonded interactions to Thr199 (Alterio et al., 2018). In addition, the ethylbenzene moiety is localized in the middle of the active site through formation of hydrophobic interactions with Leu198 present in the active site cavity. The crystal structure of CAII with Triazole, a competitive inhibitor imidazole, shows strong binding zinc(II) ion through the fourth nitrogen (Mangani and Liljas, 1993).
It is evident from the electron density map that Triazole forms two hydrogen bonds with Thr200 and Thr199 through the two adjacent nitrogen atoms (N-1 and N-2). In addition, there are several structures that report the interaction of inhibitors to the important residues of active site cavity of CAII (Andring et al., 2020; Avvaru et al., 2010; Cozier et al., 2010; Runtsch et al., 2015). Our findings are in close agreement with the reported CAII-inhibitor structures.
Pharmacokinetic properties of the selected compounds
The pharmacokinetic properties describing ADMET properties of the finally selected compounds signify their disposition in the natural environment. The criteria that influence the drug levels and kinetics of their exposure to the tissues and affect the performance and pharmacological activity can be inferred from their pharmacokinetic properties. The ADMET properties of both compounds, along with the cocrystallized inhibitor show common features with no toxic patterns as predicted from the AMES models (Table 2).
ADMET Properties of the Finally Selected Compounds Along with Known Carbonic Anhydrase II Inhibitor ZINC00638494
ADMET, absorption, distribution, metabolism, excretion, and toxicity; AMES, alternative methods for the safety evaluation of chemicals; BBB, blood brain barrier; CNS, central nervous system.
PASS analysis
The exploration of the biological activities of the selected compounds using the PASS analysis led to identifying similar types of biological properties conferring their roles in anticancer therapy. For example, the predicted properties of ZINC08918123 and ZINC00952700 are shown in Table 3. Compound ZINC08918123 and ZINC00952700 have shown their properties as cell adhesion molecule inhibitors, antineoplastic/antimutagenic, and TP53 expression enhancing potential, with the pa values ranging from 0.301 to 0.908 when pa > pi. Thus, compound ZINC08918123 possesses antineoplastic and cell adhesion molecule inhibitory potential, which could be essential to target CAII-associated diseases. At the same time, compound ZINC00952700 also shows antineoplastic potential, including MMP9 expression inhibition and TP53 expression enhancer. Therefore, the predicted biological properties of both compounds are suggesting anticancer potential.
Compounds and Their Biological Properties Calculated Through Prediction of Activity Spectra for Substances Webserver
MD simulations
Assuring the equilibration and constancy of simulated trajectories is one of the essential steps before MD analysis (Hoda et al., 2016; Naz et al., 2016, 2017, 2019). We have fetched the average potential energy for free CAII, CAII-ZINC08918123, and CAII-ZINC00952700 to determine the constancy of the simulated trajectories before MD analysis. Average potential energy for free CAII, CAII-ZINC08918123, and CAII-ZINC00952700 were—753,688, −753,713, and −753,045 kJ/mol, respectively. The enthalpy, volume, and density of all three systems suggested no significant variation, thus their equilibration during the simulation process (Table 4).
The Calculated Parameters for Carbonic Anhydrase II and Its Docked Complexes Obtained After 100 ns Molecular Dynamics Simulations
RMSD, root mean square deviation; RMSF, root mean square fluctuation; SASA, solvent accessible surface area.
Structural dynamics and residual vibrations
Any small molecule can induce significant conformational changes in protein structure after binding to its functional binding cleft (Dahiya et al., 2019; Mobley and Dill, 2009; Mohammad et al., 2020). Studying the root mean square deviation (RMSD) can help to get insights into the structural deviation and conformational changes in a protein (Kuzmanic and Zagrovic, 2010). The time evaluation of RMSD was monitored during the simulation time to investigate the stability of CAII and its complexes with ZINC08918123 and ZINC00952700. The average RMSD for CAII, CAII-ZINC08918123, and CAII-ZINC00952700 was calculated as 0.21, 0.22, and 0.20 nm, respectively (Table 4). The RMSD of all three systems measures their conformational changes over time and suggested that the simulation is in equilibrium up to 100 ns.
The RMSD plot suggests that the binding of ZINC08918123 and ZINC00952700 stabilized the CAII structure and led to fewer conformation changes (Fig. 4). A minor RMSD fluctuation appeared at 30 ns upon ZINC08918123 binding, perhaps because of its initial adjustment in the CAII-binding pocket. The binding of both compounds showed less RMSD at most of the time frames than the free form of CAII. It shows equilibration without any switching throughout the 100 ns trajectory, suggesting stability of CAII in complex with ZINC08918123 and ZINC00952700 (Fig. 4A, left panel). We also plotted the distribution of RMSD as a probability distribution function (PDF). The PDF showed that most of the complex RMSD distribution is near 0.2 nm with higher probabilities than free CAII (Fig. 4).

Structural dynamics of CAII upon compounds binding.
The RMSF analysis of the CAII backbone and its complexes with ZINC08918123 and ZINC00952700 describes local changes during the simulation. The average fluctuation for each residue was plotted to explore local structure flexibility in CAII before and after compound binding. It showed that the fluctuations of the ligand-bound complexes' residues are pretty stable compared with the apo form of CAII, especially at the site where the residues are involved in ligand binding (Fig. 4B). It indicates that the binding of ZINC08918123 and ZINC00952700 contributed to the overall structural stability of CAII.
Structural compactness
Radius of gyration (Rg) is the RMS distance of the set of atoms from their collective center of mass, which is one of the fundamental properties to be explored while studying the compactness and folding behavior of a protein (Gupta et al., 2019a, 2019b). Rg is one of the widely employed parameters to study the compactness of a protein and protein/ligand complex (Naz et al., 2018; Seeliger and De Groot, 2010). We have calculated the time evaluation of Rg from the simulated trajectory. The average of Rg for CAII-apo, CAII-ZINC08918123, and CAII-ZINC00952700 was calculated as 1.76, 1.77, and 1.76 nm, respectively. The Rg plot suggests that there was no significant change induced in the CAII packing even after ZINC08918123 and ZINC00952700 binding. Furthermore, the plot suggested that the Rg of CAII attained stable equilibrium during the 100 ns simulation (Fig. 5A).
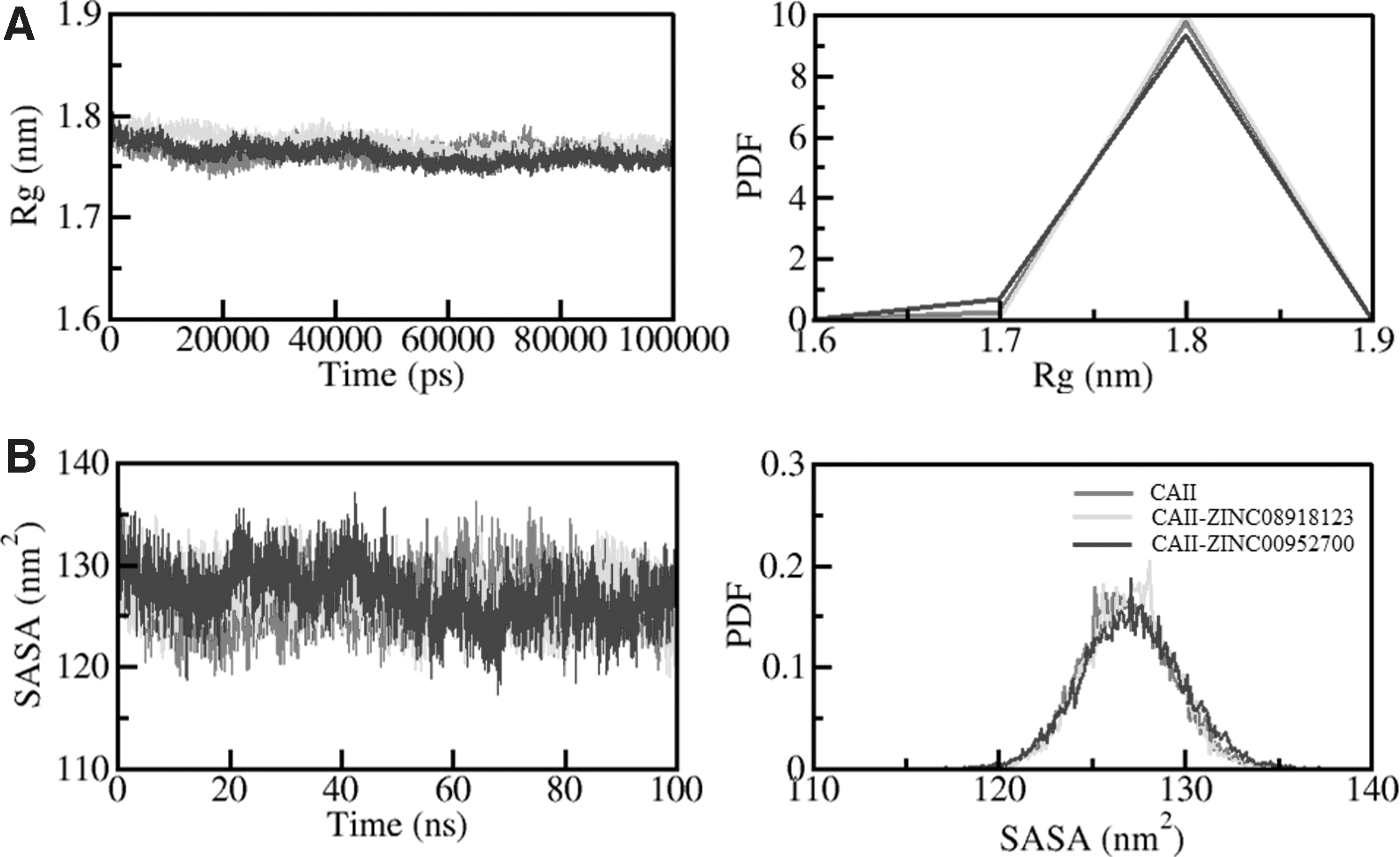
Structural compactness of CAII upon compound binding.
Solvent-accessible surface area (SASA) refers to the area of a protein molecule accessible to its adjacent solvent (Ali et al., 2014). SASA is directly associated with the Rg and is widely used to examine the conformational changes and folding/unfolding mechanism of the protein structure. While exploring the time evaluation of SASA, its average values for CAII, CAII-ZINC08918123, and CAII-ZINC00952700 complex were calculated as 126.83, 126.99, and 127.14 nm2, respectively. A minor increment in SASA values infers the exposure of some internal residues to solvent while compounds bind (Fig. 5B). There was no significant shift observed in SASA value throughout the simulation trajectory of 100 ns, thus suggesting CAII stability.
Dynamics of interactions in the CAII complexes: hydrogen bond analysis
Intramolecular H-bonding is one of the fundamental aspects of structural stability in protein (Idrees et al., 2016, 2017a, 2017b). The intramolecular H-bonding formed within 3.5 Å was calculated to explore the rearrangement of interactions in CAII during the simulation. The study further validates the stability of CAII-apo, CAII-ZINC08918123, and CAII-ZINC00952700 after the simulation. The average number of intramolecular H-bonds formed within CAII-apo, CAII-ZINC08918123, and CAII-ZINC00952700 were 186, 187, and 189, respectively (Fig. 6).

Time evolution and stability of hydrogen bonds formed intramolecularly within CAII, CAII-ZINC08918123, and CAII-ZINC00952700 complex (left panel). The probability of distribution of hydrogen bonds as PDF (right panel). PDF, probability distribution function.
Directionality and specificity of intermolecular bonding are fundamental aspects of molecular recognition in protein kinetics (Hubbard and Kamran Haider, 2001). Intermolecular H-bond analysis is widely studied to assess the constancy of interactions formed between a ligand and protein. The time evaluation of intermolecular H-bond formation during 100 ns simulation was analyzed to assess the stability of the docked complexes (Fig. 6).
The ZINC08918123-CAII complex showed up to three H-bonds, in which one was maintained for the whole simulation of 100 ns (Fig. 6A). At the same time, complex ZINC00952700-CAII showed up to four H-bonds, where two were continued in the entire simulation with some fluctuations at several time frames (Fig. 6B). H-bond analysis with MD simulation revealed that ZINC08918123 and ZINC00952700 bind to CAII with two to three H-bonds with higher fluctuations and one H-bond with the minimum fluctuation (Fig. 7).

Time evolution and stability of hydrogen bonds formed intermolecularly between
Principal component and FEL analyses
The PCA is a suitable tool to provide information about the conformational sampling of protein structure (Fatima et al., 2020). We have studied the first two PCs obtained by the PCA of CAII and its complexes with ZINC08918123 and ZINC00952700. The conformational sampling in the essential subspace of CAII-apo, CAII-ZINC08918123, and CAII-ZINC00952700 are shown in Figure 8. It confers the conformational states along the EV1 and EV2 from the Cα atom of CAII. Clustered CAII states depict that it projects a narrower range of phase spaces while in the presence of ZINC08918123 and ZINC00952700 (Fig. 8A). Furthermore, the PCA suggested that the principal motion of CAII and its complexes are higher in EV2 than EV1 (Fig. 8B). Overall, the PCA is inferring minimal conformational changes after compound binding.
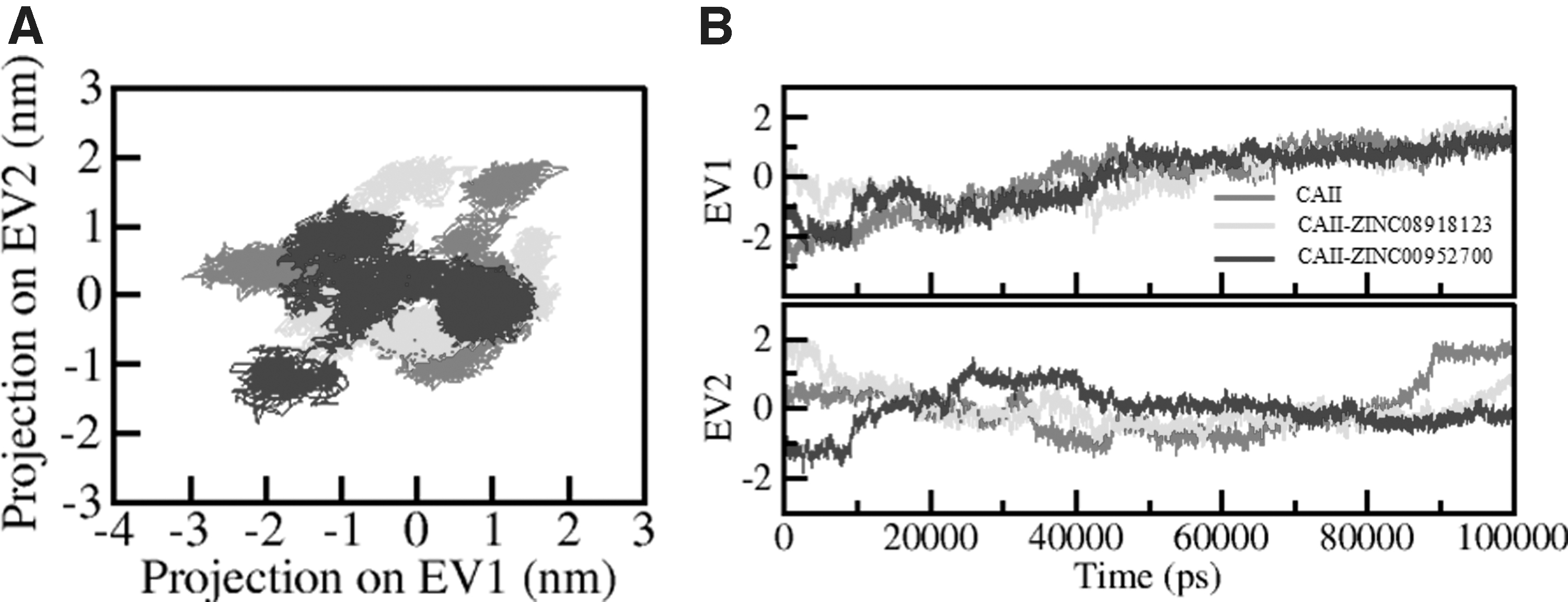
Principal component analysis.
To further understand the systems' conformational behavior and folding states, the FELs were constructed for all three systems from the simulated trajectories (Fig. 9). The FEL contour map of the free CAII portrays 2–3 stable global minima confined within majorly 3–4 basins. The deeper color indicates the lower energy, where the darkest blue represents global minima. It is found that the PC2 motion of CAII-ZINC08918123 spanned more extensive ranges than CAII and CAII-ZINC00952700 systems, indicating the conformational rearrangements caused by the ZINC08918123 binding (Fig. 9B). The CAII-ZINC00952700 showed similar basins as CAII-apo inferring complex stability during the simulation (Fig. 9C).
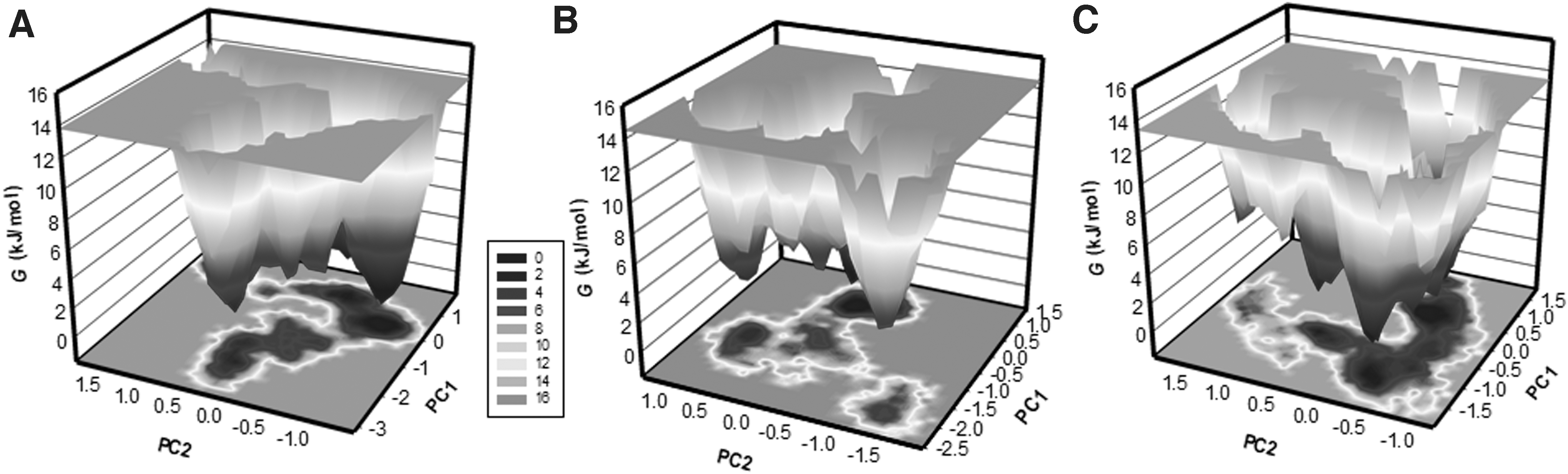
The Gibbs energy landscapes obtained during 100 ns MD simulations for
Conclusions
We present a systematic approach of vHTS to discover potential inhibitors of CAII. Initially, a library of natural products was sieved, based on the compounds' physicochemical and other drug-like properties. Then, structure-based virtual screening utilizing the molecular docking approach was performed to find high-affinity binding partners of CAII. Next, the compounds were checked for their ADMET, specific interactions, and PASS analysis to find safe and potent inhibitors of CAII.
Finally, based on the binding affinity, specific interactions, and predicted biological properties, two compounds ZINC08918123 and ZINC00952700, were chosen. We studied their potential of being therapeutic leads against cancer and glaucoma. All-atom MD simulation, PCA, and FEL studies elucidated the stable binding of ZINC08918123 and ZINC00952700 with CAII. This study used computer-assisted drug design approach and encourages the harnessing of natural products as potential CAII inhibitors in the future. Natural compounds identified in the present study specifically offer a helpful resource and strategy to discover potential promising therapeutic inhibitors of CAII to cure various cancers and glaucoma after further experimental validation and clinical studies.
Footnotes
Acknowledgments
MIH acknowledges the Indian Council of Medical Research [Grant No. ISRM/12(22)/2020] and the Council of Scientific and Industrial Research, India [Project No. 27(0368)/20/EMR-II] for financial support. The authors sincerely thank the Department of Science and Technology, Government of India for the FIST support (FIST program No. SR/FST/LSII/2020/782).
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This work was supported by Taif University Researchers Supporting Project Number (TURSP-2020/131), Taif University, Taif, Saudi Arabia.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.