
Abstract
Diabetes is a common disorder with a heterogeneous clinical presentation and an enormous burden on health care worldwide. About 1–6% of patients with diabetes suffer from maturity-onset diabetes of the young (MODY), the most common form of monogenic diabetes with autosomal dominant inheritance. MODY is genetically and clinically heterogeneous and caused by genetic variations in pancreatic β-cell development and insulin secretion. We report here new findings from targeted next-generation sequencing (NGS) of 13 MODY-related genes. A sample of 22 unrelated pediatric patients with MODY and 13 unrelated healthy controls were recruited from a Turkish population. Targeted NGS was performed with Miseq 4000 (Illumina) to identify genetic variations in 13 MODY-related genes: HNF4A, GCK, HNF1A, PDX1, HNF1B, NEUROD1, KLF11, CEL, PAX4, INS, BLK, ABCC8, and KCNJ11. The NGS data were analyzed adhering to the Genome Analysis ToolKit (GATK) best practices pipeline, and variant filtering and annotation were performed. In the patient sample, we identified 43 MODY-specific genetic variations that were not present in the control group, including 11 missense mutations and 4 synonymous mutations. Importantly, and to the best of our knowledge, the missense mutations NEUROD1 p.D202E, KFL11 p.R461Q, BLK p.G248R, and KCNJ11 p.S385F were first associated with MODY in the present study. These findings contribute to the worldwide knowledge base on MODY and molecular correlates of clinical heterogeneity in monogenic childhood diabetes. Further comparative population genetics and functional genomics studies are called for, with an eye to discovery of novel diagnostics and personalized medicine in MODY. Because MODY is often misdiagnosed as type 1 or type 2 diabetes mellitus, advances in MODY diagnostics with NGS stand to benefit diabetes overall clinical care as well.
Introduction
Diabetes is a common complex human disease with an enormous global health burden. Dissecting the molecular correlates of clinical heterogeneity of diabetes is essential for discovery of novel diagnostics and therapeutics. Maturity-onset diabetes of the young (MODY) is the most common form of monogenic diabetes with autosomal dominant inheritance. About 1–6% of patients with diabetes suffer from MODY owing to mutations in genes involved in pancreatic β-cell development and insulin secretion (Hattersley et al., 2018).
The clinical features of MODY include the presence of overt diabetes in at least three consecutive generations with autosomal dominant inheritance, absence of autoimmunity, maintenance of endogenous insulin secretion, absence of features of insulin resistance, and hyperglycemia in the absence of ketosis. These atypical features in a young patient with diabetes increase the likelihood of MODY (Gardner and Tai, 2012).
However, MODY is often misdiagnosed as type 1 diabetes mellitus (T1DM) in ∼10% of patients, or as type 2 diabetes mellitus (T2DM) in ∼5% of patients. This is because it is difficult to distinguish the clinical features that are similar and overlapping from these forms of diabetes (Covantev et al., 2016; Peixoto-Barbosa et al., 2020). Precision MODY diagnosis calls for robust biomarkers to aid clinical decision-making and personalized medicine in this important type of diabetes. A deeper understanding of MODY pathogenesis would benefit the accurate diagnosis of T1DM and T2DM by preventing misdiagnosis. Novel diagnostics would also inform prognostic assessment in the clinic for MODY and diabetes broadly (Gardner and Tai, 2012).
To date, mutations associated with MODY have been reported in 14 different genes, that is, HNF4A, GCK, HNF1A, PDX1, HNF1B, NEUROD1, KLF11, CEL, PAX4, INS, BLK, ABCC8, KCNJ11, and APPL1 (Urakami, 2019). By harnessing the targeted next-generation sequencing (NGS) approach, we report here new findings in 13 MODY-related genes, as mutations in APPL1 were discovered after the initiation of this study. The findings from the present study contribute to the worldwide knowledge base on MODY specifically, and monogenic childhood diabetes and molecular correlates of clinical heterogeneity in diabetes generally.
Materials and Methods
Characteristics of the study groups
Twenty-two patients with diabetes aged 9–18 years with the clinical suspicion of MODY and 13 healthy children and adolescent controls were included in this study (so called MODY-IST-Child) from Istanbul University, Istanbul Faculty of Medicine, Department of Pediatrics in Turkey. A group of pediatric patients (age <18 years at onset) with hyperglycemia were recruited to the MODY-IST-Child study according to at least four of the following clinical parameters:
(i) family history of diabetes in at least three generations (including the patient) suggesting an autosomal dominant inheritance, (ii) positive endogenous insulin reserve with fasting and/or stimulated C-peptide levels >0.3 and/or 0.6 ng/mL, (iii) negative pancreatic islet autoantibodies, (iv) no or low (<0.5 IU/kg/day) insulin requirement, (v) absence of ketoacidosis, (vi) absence of obesity, (vii) high-sensitive C-reactive protein (hs-CRP) level <0.7 mg/dL. Age-matched, unrelated healthy subjects without any metabolic disease were included as the control group.
A written informed consent was obtained from all patients, controls, and/or their parents. The study was approved by the Ethics Committee of Istanbul Medical Faculty, Istanbul University (dated June 20, 2014 and numbered 2014/922). Clinical parameters [age, sex, body mass index (BMI), blood pressure, medications, chronic diseases, medical interventions, and laboratory parameters] were recorded from outpatient files. This study was conducted in accordance with the Declaration of Helsinki.
DNA purification
Genomic DNA was isolated from peripheral blood samples using Epicenter MasterPure™ (Lucigen, WI, USA) DNA Purification Kit. DNA purity and concentration measurements were performed spectrophotometrically using NanoDrop 2000c (ThermoFisher Scientific, MA, USA). Extracted DNA was quantified using the Qubit™ dsDNA HS Assay Kit and the Qubit® 3.0 Fluorometer (Invitrogen, CA, USA).
Library preparation and exome sequencing
Exons and exon-intron boundaries of 13 MODY-related genes (HNF4A, GCK, HNF1A, PDX1, HNF1B, NEUROD1, KLF11, CEL, PAX4, INS, BLK, ABCC8, and KCNJ11) were amplified through long PCR using Thermal Cycler (Biorad C1000, USA). Since APPL1 has not yet been associated with MODY at the time of the study was conducted, it was not included. Libraries were constructed using the TruSeq® Custom Amplicon v1.5 Exome Library Prep kit (Illumina, Inc., San Diego, CA, USA). Paired-end sequencing of the libraries was performed on MiSeq 4000 System (Illumina, Inc.).
NGS data analysis
All samples were analyzed following the Genome Analysis ToolKit (GATK) Best Practices Pipeline (Van der Auwera et al., 2013) using Trimmomatic v0.27 (Bolger et al., 2014) (trimming), Burrows-Wheeler Aligner v.0.7.12 (Li and Durbin, 2009) (alignment), GATK IndelRealigner v3.3.0 (local alignment), and GATK Unified Genotyper (DePristo et al., 2011) [detection of single-nucleotide polymorphisms (SNPs) and in/del mutations]. The human genome build37 (GRCh37/hg19) was used as the reference genome (http://genomereference.org).
Considering the allelic frequency data obtained from 1000 Genomes phase 3 (build 2013-05-02) (The 1000 Genomes Project Consortium, 2012) and Exome Aggregation Consortium (ExAC) v3.1 (Karczewski et al., 2020), variants with an overall population frequency of <5% were filtered and compared among study groups.
Variant annotation was performed using The Single Nucleotide Polymorphism Database (dbSNP) build 146 (Kitts and Sherry, 2002), ClinVar (Landrum et al., 2020), and dbNSFP v3.0 (Liu et al., 2011, 2016). For the identified missense mutations, SIFT (Kumar et al., 2009) and PolyPhen-2 (Adzhubei et al., 2010) were used to assess the likelihood of pathogenic effect. The status, novelty, functional consequence, and pathogenicity of the variants were also investigated via databases, including OMIM (https://omim.org/) (McKusick, 2007), NCBI-ClinVar (Landrum et al., 2020), human phenotype ontology (Köhler et al., 2021), and Mutation Assessor (Reva et al., 2011).
Minor allele frequency (MAF) of variants were primarily based on TOPMED (www.nhlbiwgs.org; NHLBI [National Heart, Lung and Blood Institute], 2014) and gnomAD v3.1 (Karczewski et al., 2020) projects; in the lack of data, MAF calculations were based on the present study. HWE.test and HWE.chisq functions of the Genetics v1.3.8.1 (Warnes et al., 2013) R package was used to estimate disequilibrium and test for Hardy-Weinberg equilibrium (HWE). The relative risks (RR) were determined and visualized using R packages cBioPortalData v2.2.8 (Ramos et al., 2020) and forest plot v1.10.1 (https://cran.r-project.org/web/packages/forestplot). The MutationMapper tool of cBioPortal was used to map mutations on proteins and their domains (Gao et al., 2013).
Before molecular diagnosis, the MODY probability calculator (MPC) scores were calculated in all cases with clinically suspected MODY through MPC (v.1.0) provided by The Exeter Diabetes App (Shields et al., 2012).
Statistical analysis
The statistical analyses were performed using the SPSS v.20.0 (IBM SPSS, Inc., Chicago, IL, USA). Quantitative variables were represented as means and standard deviations, and Student's t-test was used for comparison between two groups. The categorical variables were presented as numbers and percentages and the statistical analyses between study groups were performed using the chi-square test. p-values <0.05 were considered statistically significant.
Results
Characterization of the patients enrolled in the study
When demographic, clinical, and biochemical characteristics of the study groups are comparatively analyzed (Table 1), patients with MODY (n = 22) and healthy controls (n = 13) displayed a similar distribution of gender and age (p > 0.05). Although serum fasting lipid profile, thyroid-stimulating hormone, free thyroxine, creatinine, and hemogram parameters did not differ statistically (p > 0.05), diastolic blood pressure (p = 0.043) and fasting blood glucose (FBG) levels were higher in the MODY group compared to the control group, as expected (p < 0.001).
Characteristics of the Study Population
BMI, body mass index; DBP, diastolic blood pressure; FT4, free thyroxine; HbA1c, hemoglobin A1c; HDL-C, high-density lipoprotein cholesterol; Hgb, hemoglobin; hs-CRP, high-sensitive C-reactive protein; Htc, hematocrit; LDL-C, low-density lipoprotein cholesterol; MODY, maturity-onset diabetes of the young; SBP, systolic blood pressure; TG, triglycerides; Total C, total cholesterol; TSH, thyroid-stimulating hormone; VLDL-C, very low-density lipoprotein cholesterol.
Boldface = statistically significant.
Targeted sequencing and identification of genetic mutations
Paired-end sequencing (with an average length of 150 bp) of the exons and exon-intron boundaries (±100 bp) of 13 MODY-related genes resulted in an average sequencing depth of 221,000 high-quality reads per sample and 35 Mb of data per sample. Following the GATK best practices pipeline and using the human genome build37 (GRCh37/hg19) as the reference genome, we identified 133 genetic variations in 13 MODY-related genes (Supplementary Figs. S1–S13).
We observed that the mutation loads varied among the MODY. For example, the highest number of mutations was observed in the ABCC8 gene (n = 33) followed by HNF1A (n = 21), BLK (n = 17), CEL (n = 13), and HNF1B (n = 10) genes. Six variations were detected in each of the HNF4A, KCNJ11, PAX4, and PDX1 genes. The fewer variations were identified in the genes GCK (n = 5), KLF11 (n = 3), NEUROD1 (n = 3), INS (n = 2), and PDX1-AS1 (n = 2).
MODY-specific genetic mutations
We identified 43 MODY-specific mutations that were observed in MODY patients but not in the control group. These mutations were annotated, and their status, novelty, functional consequence, genomic features, and pathogenicity were further investigated (Table 2).
Genetic Variations Specific to Pediatric Patients with Maturity-Onset Diabetes of the Young
CI, confidence interval; HWE, Hardy-Weinberg equilibrium; MAF, minor allele frequency; UTR, untranslated region.
Annotation of MODY-specific variants revealed 11 missense mutations in 9 genes (GCK p.L315F; HNF1A p.R272G; NEUROD1 p.D202E; NEUROD1 p.P197H; KLF11 p.R461Q; CEL p.S712P; PAX4 p.R164Q; BLK p. G248R; BLK p.P39K; ABCC8 p.C418R; and KCNJ11 p.S385F) (Table 3) in addition to 4 synonymous mutations, 23 intron variants, 3 untranslated region (UTR) variants, and 2 noncoding ribonucleic acid (ncRNA) variants in various genes (Table 4).
Missense Mutations Specific to Pediatric Patients with Maturity-Onset Diabetes of the Young
dbSNP, The Single Nucleotide Polymorphism Database; RS, reference SNP.
Synonymous and Noncoding Sequence Variations in the Pediatric Patients with Maturity-Onset Diabetes of the Young
ncRNA, noncoding ribonucleic acid.
When analyzed based on the MODY subtype, we made the following observations:
Three HNF4A variations (one intronic, one synonymous, and one 3′UTR) were detected in MODY1, Two GCK variations (one intronic and one missense) in MODY2, Five HNF1A variations (three intronic, one missense, and one 3′UTR) in MODY3, Two PDX1 ncRNA variants in MODY4, Three HNF1B (two intronic and one synonymous) in MODY5, Two NEUROD1 missense mutations in MODY6, One KLF11 missense mutation in MODY7, Seven CEL variations (five intronic, one synonymous, and one missense) in MODY8, Three PAX4 variations (two intronic and one missense) in MODY9, Eight BLK variations (four intronic, two missense, one synonymous, and one 3′UTR) in MODY11, Six ABCC8 variations (five intronic and one missense) in MODY12, and One KCNJ11 missense mutation in MODY13.
However, no variation was detected for the INS gene, that is, in MODY10.
The RRs changed in the range of 1.62–1.87 for all variants, suggesting that MODY risk increased with the presence of variation. In addition, violations of HWE assumptions were observed for the majority of MODY-specific variants (p > 0.05) except for two (an ncRNA variant in PDX1-AS1 and an intron variant in HNF1B) (Table 2).
Among the missense variations, 5 mutations were already associated with MODY subtypes in ClinVar, and the majority (8 out of 11) were presented in the dbSNP database, except, notably, 3 variations (HNF1A p.R272G, NEUROD1 p.D202E, and CEL p.S712P). On the contrary, four missense mutations (NEUROD1 p.D202E, KFL11 p.R461Q, BLK p.G248R, and KCNJ11 p.S385F) were associated with MODY subtypes for the first time in the present study (Table 3).
The likelihood of pathogenicity was predicted as deleterious in eight variations (GCK p.L315F, HNF1A p.R272G, NEUROD1 p.P197H, KLF11 p.R461Q, PAX4 p.R164Q, BLK p.P39K, BLK p.G248R, and ABCC8 p.C418R) and tolerated in two variations (NEUROD1 p.D202E and CEL p.S712P), while no prediction could be achieved for KCNJ11 p.S385F (Table 3).
Among the missense mutations predicted to be deleterious were two variations in the homeobox transcription factor KN domains of the Hnf1A (p.R272G) and Pax4 (p.R164Q) proteins, one variation (p.L315F) in the hexokinase two domain of the Gck protein, and one variation (p.P197H) in the neuronal helix-loop-helix transcription factor domain of the NeuroD1 protein (Fig. 1). Moreover, a variation (p.R461Q) in the C4-type zinc finger domain of the Klf11 protein, a variation (p.G248R) in the protein tyrosine kinase domain of the Blk protein, and a variation (p.C418R) in the ABC-transporter transmembrane region of the Abcc8 protein were also predicted to be deleterious (Fig. 2).
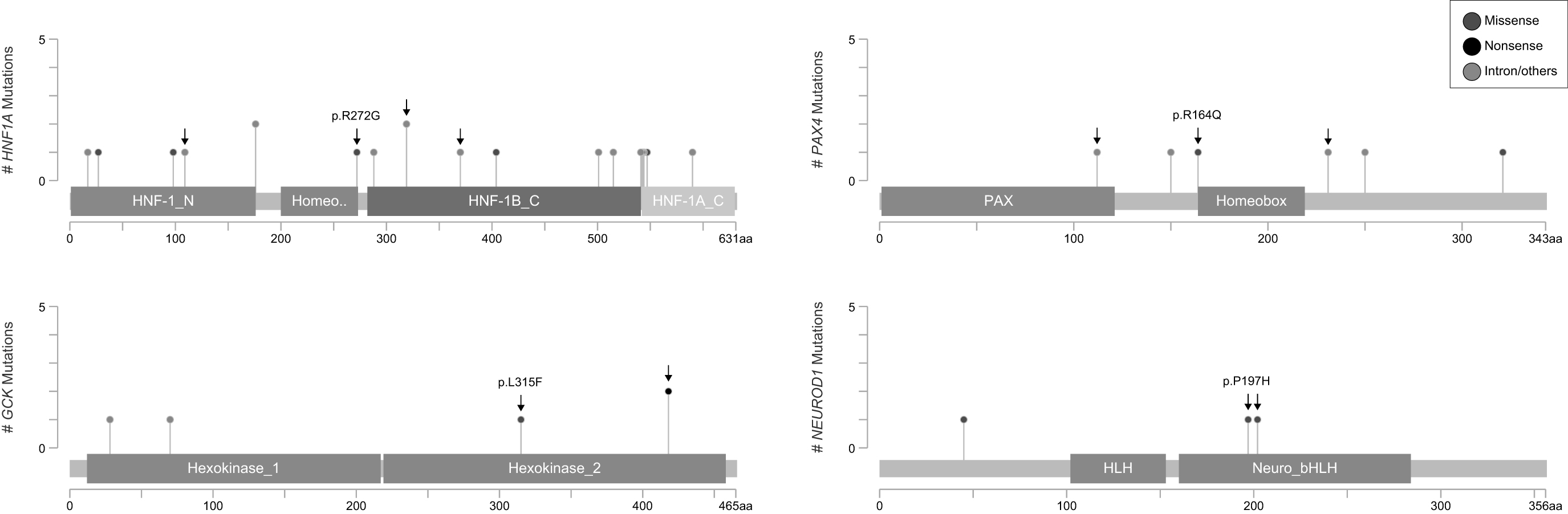
Missense mutations in HNF1A, PAX4, GCK, and NEUROD1 genes identified using next-generation sequencing in the present study. The novel mutations were marked with an arrow, and the deleterious mutations were highlighted indicating their identifier.

Missense mutations in KLF11, BLK, ABCC8, and CEL genes identified using next-generation sequencing in the present study. The novel mutations were marked with an arrow, and the deleterious mutations were highlighted indicating their identifier.
Three of the identified synonymous variations were already presented in the dbSNP database and two of them were already associated with MODY subtypes. On the contrary, a variant (p.S367) in the HNF1B gene was detected in MODY patients for the first time in this study (Table 4).
The majority of the identified intron variants was already presented in the dbSNP database, except 3 out of 23 (Table 4). However, 15 out of 23 variations were not previously detected in MODY patients and were not associated with MODY subtypes in the literature. Similarly, the identified UTR and ncRNA variants were associated with MODY patients for the first time in this study, except the 3′UTR variant in HNF4A.
Discussion
This study offers new insights on molecular correlates of MODY and has both molecular and clinical ramifications to improve clinical diagnosis and prevent misdiagnosis in the future (Table 5). While the findings call for future functional genomics and comparative population genomics studies, the novel genetic variants identified herein using the targeted NGS experimental approach adds to the body of knowledge base on MODY specifically, and diabetes care generally.
Clinical and Biochemical Characteristics of Patients with Identified Maturity-Onset Diabetes of the Young Mutations
FBG, fasting blood glucose; MPC, MODY probability calculator; SU, sulfonylurea; T1DM, type 1 diabetes mellitus.
Boldface = missense mutations.
Targeted sequencing of exons and exon-intron boundaries revealed that genetic variation was unevenly distributed across genes. Our attention was mainly focused on missense mutations in MODY genes, as the resulting amino acid change can alter protein function. In addition to mutations already associated with MODY subtypes in the literature, in the present study, we discovered, for the first time, 4 missense mutations (NEUROD1 p.D202E, KFL11 p.R461Q, BLK p.G248R, and KCNJ11 p.S385F), 1 synonymous mutation (HNF1B p.S367), and 15 intron variants, 2 UTR, and 2 ncRNA variants in MODY patients. Two novel missense mutations (i.e., KLF11 p.R461Q and BLK p.G248R), localized to the C4-type zinc finger and protein tyrosine kinase domains of the proteins, respectively, were predicted to be deleterious, suggesting potentially significant relevance for further studies.
Contextualizing the findings in MODY-related genes
HNF4A-MODY1
The transcription factor Hnf4A regulates direct insulin expression and the expression of genes involved in glucose transport and metabolism, particularly the major glucose transporter, GLUT2 (Stoffel and Duncan, 1997). Therefore, mutations in the HNF4A gene lead to progressive failure of insulin secretion and deterioration of glucose tolerance with age. Heterozygous HNF4A mutations have effects on glucose metabolism due to the β-cell dysfunction and lead to macrosomia and hyperinsulinemic hypoglycemia in infancy (Pearson et al., 2007), or MODY1 in adulthood (Nkonge et al., 2020; Yamagata et al., 1996). Low levels of high-density lipoprotein cholesterol (HDL-C) and triglycerides, and elevated low-density lipoprotein cholesterol (LDL-C) are the basic characteristics of MODY1 patients (McDonald and Ellard, 2013; Nkonge et al., 2020).
In the present study, three HNF4A variations (one intronic, one synonymous, and one 3′UTR) were detected. The synonymous mutation (p.N379) and the 3′UTR variant (c.*76G>A) have already been associated with MODY1 in the literature and presented in ClinVar. However, there are no studies in the literature on their phenotypic effects. In addition, this study was the first to identify the intron variant (c.50-3351C>A) in MODY patients. The patient who carried the HNF4A p.N379 synonymous mutation also carried the missense mutations GCK p.L315F and KLF11 p.R461Q.
GCK-MODY2
While homozygous inactivating mutations of the GCK gene lead to complete Gck deficiency and eventually to neonatal diabetes, inactivating heterozygous mutations in the gene encoding Gck, which functions as a “glucose sensor” in β-cells, resulting in a partial deficiency of the enzyme and development of MODY2 (García-Herrero et al., 2007; Gloyn, 2003). MODY2 is characterized by mild fasting hyperglycemia from birth and minor deterioration in glycemia with increasing age (Chakera et al., 2015; Ozdemir, 2018). MODY2 patients are diagnosed incidentally during a routine examination and usually have normal hemoglobin A1c (HbA1c) and measurable C-peptide levels, or rarely, they may be associated with gestational diabetes mellitus (GDM) (McDonald and Ellard, 2013; Osbak et al., 2009; Rudland et al., 2016).
In the present study, two GCK variations (one intronic and one missense) were detected. This study was the first to identify the intronic variant (c.1253 + 49G>A) in MODY patients. The missense variant (p.L315F) was previously detected in the Belgian, Luxembourgian, and Turkish population (Aykut et al., 2018; Vits et al., 2006; Yilmaz-Agladioglu et al., 2016) and has been associated with MODY2. The deleterious p.L315F mutation is located on the hexokinase two domain of the Gck protein, the catalytic subunit of the enzyme that converts glucose to glucose-6-phosphate in an ATP-dependent manner. The male patient with p.L315F mutation (patient code: CH9) also carried heterozygous KLF11 p.R461Q missense mutation. The patient received no antidiabetic treatment, had high FBG levels, and very low insulin/C-peptide levels. His clinical features support a double heterozygosity for MODY2/MODY7.
HNF1A-MODY3
MODY3 is associated with progressive and worsening glycemic control with age and micro and macrovascular complications similar to T1DM and T2DM. The transcription factor Hnf1A regulates the expression of the INS gene in pancreatic β-cells (Dukes et al., 1998; Pontoglio et al., 1998). The promoter of the CRP gene has the binding site for the Hnf1A. HNF1A mutations that disrupt the structure of the CRP binding site of the transcription factor can also reduce Crp expression. Therefore, a low hs-CRP level is a useful biomarker for the differential diagnosis of HNF1A-MODY (Gardner and Tai, 2012; Owen, 2013). In addition, a higher HDL-C level compared to T2DM and close to controls is helpful in the clinical differential diagnosis of HNF1A-MODY (McDonald and Ellard, 2013).
Heterozygous mutations in the HNF1A cause progressive insulin deficiency leading to mild hyperglycemia in childhood and diabetes in early adulthood (Hattersley et al., 2018; Hwang et al., 2006). More than 400 different HNF1A mutations have been identified (Colclough et al., 2013; Ellard and Colclough, 2006). The prevalence of HNF1A-MODY is high in European, North American, and Asian population (Hattersley et al., 2018; Hwang et al., 2006; Kavvoura and Owen, 2013; Shields et al., 2010).
We identified five HNF1A variations (three intronic, one missense, and one 3′UTR) in the MODY3 subtype in this study. The missense mutation (p.R272G) located on the KN domain of Hnf1A was previously detected in children and adolescents with MODY (Goksen et al., 2013). Two of the three intronic variants found in Japanese and Portuguese populations were associated with T2DM (Imamura, 2016; Mafra, 2017); however, the novel intronic variant (c.326 + 45G>C) identified in this study was associated with MODY for the first time.
Moreover, the 3′UTR variant (c.*87G>A) is also associated with MODY in this study. The patient with the HNF1A R272 mutation was a 14-year-old girl (patient code: CH4) receiving insulin therapy. The patient had high FBG and HbA1c levels but low triglycerides levels. This patient's 16-year-old brother (patient code: CH5) had the 3′UTR c.*87G>A variant in HNF1A. Similar to his sister, he was compatible with the HNF1A-MODY phenotype in terms of clinical and biochemical findings. Differently, his hs-CRP level was low. The MPC scores of these siblings were 75.5%.
PDX1-MODY4
Pdx1 (Ipf1) plays an important role in pancreatic development and β-cell function by stimulating the expression of the genes INS, GLUT2, and GCK (Stoffers et al., 1997). Homozygous mutations of the PDX1 gene cause pancreatic agenesis and neonatal diabetes, while heterozygous mutations (MODY4) lead to β-cell dysfunction and hyperglycemia (Sanyoura et al., 2018). PDX-1 mutations may also play a role in GDM susceptibility (Gragnoli et al., 2005). Previous studies have also reported that PDX1 gene mutations can lead to decreased protein expression and T2DM (Macfarlane et al., 1999; Reis et al., 2000).
In the present study, we did not observe any mutation in the PDX-1 gene; but identified two novel ncRNA variations (c.-58G>A and c.-51A>G) in PDX1-AS1 (also known as PLUTO or HI-LNC71), the antisense transcript long ncRNA located 3 kb upstream of the PDX1 gene. It has been shown that decreased PDX1-AS1 expression in T2DM patients leads to a significant decrease in PDX1 expression by reducing the interaction of the PDX1 promoter with its enhancer (Akerman et al., 2017; Das et al., 2018). The c.-58G>A variant was detected in eight patients (five heterozygous, three homozygous), and the c.-51A>G variant was found heterozygous in one patient who also carried the c.-58G>A variant. Our findings confirm the role of ncRNAs in the regulation of β-cell development and function, and we suggest that ncRNA variants of PDX-AS1 may also be effective in the development of MODY.
HNF1B-MODY5
Hnf1B (Tcf2) has been shown to function in nephron development and regulates organogenesis of the kidney, pancreas, genitourinary system, liver, lung, and intestine (Coffinier et al., 1999). Heterozygous mutations in the HNF1B gene have been shown to cause renal cysts and diabetes syndrome, genitourinary tract malformations, liver dysfunction, and thyroid abnormalities as well as hepatic insulin resistance and β-cell dysfunction (Bellanné-Chantelot et al., 2005; Edghill et al., 2006; Lim et al., 2020). Renal dysfunction in MODY5 patients usually occurs in childhood and diabetes develops in adolescence or early adulthood (Bellanné-Chantelot et al., 2004). Measurable C-peptide levels, elevated creatinine and liver enzymes, hypomagnesemia and hyperuricemia are major biochemical properties of MODY5 (McDonald and Ellard, 2013).
Here, we identified a novel synonymous mutation (p.S367) in addition to the two intronic mutations (c.1535-23C>T and c.1535-48C>T) in the HNF1B gene. The patient with the synonymous mutation (p.S367) was an 11-year-old girl (patient code: CH2) who did not receive any antidiabetic therapy. In the present case, the age at onset of diabetes, thyroid dysfunction, a measurable C-peptide level, absence of autoantibodies to pancreatic islets, and a novel mutation in HNF1B indicated MODY5. The intronic variations were presented in dbSNP, but none of the three variations was previously associated with MODY.
NEUROD1-MODY6
The nuclear transcription factor NeuroD1 is expressed in pancreatic and neuronal cells and is involved in pancreatic β-cell development, insulin expression, and regulation of neuronal development (Horikawa and Enya, 2019). Heterozygous mutations in NEUROD1 cause β-cell dysfunction and eventually diabetes beginning in childhood or adulthood (MODY6), while homozygous mutations can lead to neonatal diabetes and neurological abnormalities (Oliveira et al., 2020).
In this study, p.D202E and p.P197H missense mutations were detected in the NEUROD1 gene and the latter was described also as an SNP (Malecki et al., 1999). Both mutations were located in the neuronal helix-loop-helix transcription factor domain of the NeuroD1 protein. The p.P197H mutation was predicted to be deleterious, whereas p.D202E was predicted to be benign. The p.P197H mutation was found in four patients in our study group (patient codes; CH5, CH17, CH21, and CH23), but none of them was diagnosed with a neurological disorder. The patients with the p.P197H mutation were diagnosed with MODY3 and MODY11, respectively, because one of them had HNF1A (patient code: CH5) and the other had BLK gene mutations (patient code: CH23) and their clinical phenotype was consistent with these MODY subtypes.
There were only two patients with NEUROD1 p.P197H, but no other MODY mutations (patient codes; CH17 and CH21). One of the patients was a 16-year-old girl on insulin therapy and her MPC score (0.7%) and C-peptide level were very low. The patient with high glucose and cholesterol levels was evaluated as MODY6. The other patient was a 10-year-old girl on metformin (patient code: CH17) with a MPC score of 75.5%. Biochemical findings of this patient were within normal limits. These two patients with only NEUROD1 p.P197H mutation were diagnosed with MODY6.
The p.D202E mutation, on the contrary, is novel; it has not been previously presented in ClinVar and has not been associated with MODY. The patient with the p.D202E mutation (patient code: CH18) also had the p.S712P missense mutation (likely benign) at the CEL gene. However, this patient had a very low MPC score (0.7%). When evaluated together with biochemical findings (high FBG and HbA1c levels, and a low C-peptide level), the benign NEUROD1 p.D202E and CEL p.S712P missense mutations are unlikely to be associated with the development of MODY, thus, this patient was found to be more likely to have T1DM.
KLF11-MODY7
Klf11 is a zinc finger nuclear transcription factor expressed in pancreatic islets and functions as a glucose-induced regulator of the INS gene (Fernandez-Zapico et al., 2009). Heterozygous mutations (MODY7) in the KLF11 gene cause β-cell dysfunction scavengers and impaired insulin secretion by modulating the expression of free radical scavengers (Jang, 2020; Lomberk et al., 2013; Nkonge et al., 2020).
In the present study, we identified a novel missense mutation (p.R461Q) in a male patient (patient code: CH9). He also had a synonymous mutation (p.N379) in HNF4A and a missense mutation (p.L315F) in GCK. The KLF11 p.R461Q was localized to the C4-type zinc finger domain of the Klf11 protein and was predicted to be deleterious.
CEL-MODY8
The lipase Cel is sent into the intestinal lumen along with pancreatic digestive enzymes to participate in the hydrolysis of dietary lipids (Raeder et al., 2006). Heterozygous mutations in the CEL gene (MODY8) are associated with pancreatic atrophy, exocrine and endocrine pancreatic dysfunction, and diabetes (Torsvik et al., 2010). MODY8 is thought to be a protein misfolding disease resulting from negative gain of function of the mutant proteins in the pancreas (Johansson et al., 2011).
We identified seven MODY-related variants (five intronic, one synonymous, and one missense) in the CEL gene. Among these, the benign missense mutation (p.S712P), the synonymous mutation (c.450C>T), and four of the intronic variants have been identified in ClinVar. However, the intronic variant (c.76-29G>A) was predicted for the first time in the present study in MODY patients.
PAX4-MODY9
The nuclear transcription factor Pax4 has functions in fetal development, regulation of β-cell differentiation, and repression of the activity of the promoters of INS and glucagon (Smith et al., 1999). Heterozygous mutations in the PAX4 gene (MODY9) cause abnormal β-cell development and result in β-cell dysfunction and impaired glucose-dependent insulin secretion (Plengvidhya et al., 2007; Sujjitjoon et al., 2016). MODY9 has been associated with ketosis-prone diabetes (Mauvais-Jarvis et al., 2004).
In the current study, we identified three variations (two intronic and one missense) in PAX4. The missense mutation (p.R164Q) was detected in a 7-year-old girl in a heterozygous state (patient code: CH12) in our study. The clinical phenotype of this girl who did not receive any antidiabetic therapy was consistent with PAX4-MODY and was diagnosed as MODY9. The mutation was localized in the KN domain of the protein and predicted to be deleterious. The arginine amino acid at the PAX4 homeodomain position is an evolutionarily conserved residue in several species. The conversion of the amino acid from positively charged arginine to uncharged/polar glutamine at this position has been reported to have deleterious effects (Abreu et al., 2020).
INS-MODY10
The gene INS encodes the precursor of the insulin (preproinsulin). MODY10 is characterized by reduced β-cell mass and insulin secretion, and variable onset diabetes (Boesgaard et al., 2010; Edghill et al., 2008). No mutation in the INS gene was reported in our study in patients clinically diagnosed with MODY.
BLK-MODY11
BLK is a nonreceptor tyrosine kinase that belongs to the Src proto-oncogene family and is expressed in β-cells. It promotes glucose-dependent insulin synthesis and secretion by upregulating the transcription factors PDX-1 and NKx-6 (Nkonge et al., 2020). Heterozygous mutations in the BLK gene (MODY11) that decrease BLK expression and/or activity lead to decreased insulin secretion and ultimately diabetes. BLK mutations associated with MODY11 cause a very rare MODY subtype. MODY11 is associated with a higher prevalence of the obese phenotype compared to other MODY subtypes (Borowiec et al., 2009).
We identified eight BLK variations (four intronic, two missense, one synonymous, and one 3′UTR) in MODY patients. The p.P39K and p.G248R missense mutations were predicted to have damaging effects on Blk protein structure/function, and no previous studies have reported these mutations. The p.P39K and synonymous mutation p.P237 are described as benign in ClinVar, but p.G248R is not defined in ClinVar.
In all three patients with BLK mutations, the MPC score was 75.5%. A 13-year-old female patient (patient code: CH1) with BLK p.P39K missense mutation also had PDX1AS1 c.-58G>A and c.-51A>G ncRNA variants heterozygous. In this patient who did not receive treatment, insulin level was quite high, but FBG and BMI values were in the normal range, which is not consistent with the BLK-MODY phenotype. However, BLK-MODY is known to have incomplete penetration and not all carriers have diabetes (Delvecchio et al., 2020). It has been reported that environmental and genetic factors play an important role in the development of BLK-MODY. Previous studies associated with MODY11 have suggested that BLK mutations may be “diabetogenic” through obesity-related mechanisms (Bonnefond et al., 2013; Borowiec et al., 2009). Therefore, we cannot exclude that the effect of BLK p.P39K mutation on clinical phenotype may not have occurred due to the absence of obesity in this patient.
The synonymous BLK p.P237 mutation was detected in an 11-year-old boy (patient code: CH3) who had no mutations in other MODY genes. This patient who did not receive antidiabetic treatment was hyperglycemic was compatible with the BLK-MODY phenotype, except for obesity. On the contrary, the deleterious p.G248R mutation localized to the protein tyrosine kinase domain of the BLK protein was newly detected in a Turkish MODY patient. The novel p.G248R missense mutation was discovered in a 12-year-old boy (patient code: CH23) who also had the NEUROD1 p.P197H mutation. The clinical features of this patient supported the BLK-MODY phenotype.
ABCC8-MODY12 and KCNJ11-MODY13
The ATP-sensitive potassium channel (K-ATP) in the β-cell membrane has a function in the regulation of glucose-stimulated direct insulin secretion. The ABCC8 gene encodes the sulfonylurea (SU) receptor 1 subunit of K-ATP (Bonnefond et al., 2012; Gloyn et al., 2004; Vaxillaire et al., 2004). The KCNJ11 gene encodes the Kir6.2 subunit of the K-ATP channel (Sakura, et al., 1996). Heterozygous mutations of the ABCC8 (MODY12) and KCNJ11 (MODY13) genes result in dysfunction of subunit interactions in the potassium channel and impaired insulin secretion and glucose intolerance. Among the very rare MODY subtypes, MODY12 and MODY13 are associated with SU-sensitive diabetes (De Franco et al., 2020) and may be characterized by congenital hypoglycemic hyperinsulinemia or adult-onset diabetes (Nkonge et al., 2020).
In the present study, we detected six variations (five intronic and one missense) in ABCC8 and one novel missense mutation in KCNJ11. The missense mutation (p.C418R) in ABCC8 (patient code: CH20) and the missense mutation (p.S385F) in KCJN11 (patient code: CH15) were detected in one patient each. Both patients had low C-peptide levels and their clinical features supported the MODY12 and MODY13 subtypes according to their mutation. The p.C418R mutation, localized in the ABC-transporter transmembrane region of the ABCC8 protein, was previously reported at a frequency of 0.06% (n = 5) in the epidemiological DESIR study conducted in over 4000 normoglycemic subjects; however, the authors claimed that it may not be associated with diabetes (Vaxillaire et al., 2008). To our knowledge, the KCJN11 mutation p.S385F was found for the first time in this study.
Conclusions
When the clinical and biochemical findings of the patients are evaluated together with the molecular and bioinformatics results, MODY-associated mutations were detected in 12 of the patients diagnosed with MODY: 5 novel (4 missense mutations—NEUROD1 p.D202E, KFL11 p.R461Q, BLK p.G248R, and KCNJ11 p.S385F, a synonymous mutation—HNF1B p.S367, a 3′UTR variant—HNF1A c.*87G>A), and 8 previously described mutations (GCK p.L315F, HNF1A p.P272G, NEUROD1 p.P197H, PAX4 p.R164Q, BLK p.P39K, BLK p.P237, KCJN11 p.S385F, and ABCC8 p.C418R). While seven patients with clinical features of MODY were diagnosed as “MODYX” due to the absence of a causative gene mutation, and three patients were evaluated as type 1 diabetes.
Taken together, this study provides novel insights into the molecular and clinical features of 13 MODY subtypes in pediatric patients with clinical suspicion of MODY. While the present study sample size is limited and thus calls for future studies in larger samples from diverse world populations, the NGS utilized herein offers deeper molecular insights than conventional molecular approaches.
We think that the novel mutations identified in this study contribute to a comprehensive and bigger picture on the genetic underpinnings of MODY. Most importantly, this study attests to the fact that the diagnosis of MODY calls for an interdisciplinary approach that builds on and integrates evidentiary streams from astute clinical observations, DNA sequencing, and bioinformatics data. In this sense, MODY is ideally suited for integrative biology driven diagnostics and therapeutics in the future. Since MODY is often misdiagnosed as T1DM or T2DM, advances in MODY diagnostics with NGS stand to benefit diabetes overall clinical care as well.
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
The present study was supported by the intramural research fund of the Istanbul University (Project No. 44381). The scholarships under the TUBITAK 2211-A and YOK 100/2000 Doctoral Fellowship Programs provided to Gizem Gulfidan are greatly acknowledged.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.