
Abstract
Gastric cancer (GC) is a prevalent disease worldwide with high mortality and poor treatment success. Early diagnosis of GC and forecasting of its prognosis with the use of biomarkers are directly relevant to achieve both personalized/precision medicine and innovation in cancer therapeutics. Gene expression signatures offer one of the promising avenues of research in this regard, as well as guiding drug repurposing analyses in cancers. Using publicly accessible gene expression datasets from the Gene Expression Omnibus and The Cancer Genome Atlas (TCGA), we report here original findings on co-expressed gene modules that are differentially expressed between 133 GC samples and 46 normal tissues, and thus hold potential for novel diagnostic candidates for GC. Furthermore, we found two co-expressed gene modules were significantly associated with poor survival outcomes revealed by survival analysis of the RNA-Seq TCGA datasets. We identified STAT6 (signal transducer and activator of transcription 6) as a key regulator of the identified gene modules. Finally, potential therapeutic drugs that may target and reverse the expression of the identified altered gene modules examined for drug repurposing analyses and the unraveled compounds were further investigated in the literature by the text mining method. Accordingly, we found several repurposed drug candidates, including Trichostatin A, Vorinostat, Parthenolide, Panobinostat, Brefeldin A, Belinostat, and Danusertib. Through text mining analysis and literature search validation, Belinostat and Danusertib were suggested as possible novel drug candidates for GC treatment. These findings collectively inform multiple aspects of GC medical management, including its precision diagnosis, forecasting of possible outcomes, and drug repurposing for innovation in GC medicines in the future.
Introduction
Gastric cancer (GC) is one of the prevalent malignant cancers in the world (Shi et al., 2014). The development and progression of GC initiate in epithelial cells of stomach mucosa (Shi et al., 2014). In 2018, it was reported that there were about 1,000,000 new GC cases and 783,000 deaths wherein GC led as the fifth highest prevalent cancer type (Bray et al., 2018). The prognostic outcome is very low since most GC cases are diagnosed in the advanced stage of the tumor, making the treatment opportunities limited (Van Cutsem et al., 2016). Therefore, detection of a prognostic gene signature for patients with GC may improve the patients' survival outcome.
Recently, network-based approaches have been utilized to provide context-specific network models for human complex diseases and several studies have been reported from our research group, particularly in ovarian cancer (Gov, 2020; Gov and Arga, 2017). Analyses of differential co-expression networks by topological assessment offers valuable knowledge regarding biological alterations based on gene expression by comparing two context-specific conditions, such as diseased tissues and healthy tissues (Gov, 2020; Hsu et al., 2015; Ideker and Krogan, 2012; Kori et al., 2019).
Systems biology can be defined as effective collaborative ways in drug discovery and development using interactions between different system components (Butcher et al., 2004). Previously systems biology-based drug repurposing analyses were conducted in several diseases such as osteosarcoma (Andrade et al., 2021), colorectal cancer (Beklen et al., 2021; Irham et al., 2020), and COVID-19 (Auwul et al., 2021, p. 19).
Liu et al. (2018) analyzed several microarray datasets of GC from a public database and demonstrated hub genes and a prognostic gene signature. Similar studies also investigated the GC datasets and proposed gene signatures and potential gene-based biomarkers that may be utilized for early diagnosis and prognosis prediction utilizing microarray datasets (Chia and Tan, 2016; Lei et al., 2013; Tan et al., 2011). Despite the many studies developed to decode the molecular signatures of GC through systems biology approaches, the identification of effective therapeutic agents has remained a challenge.
Our approach is significantly different from the previous studies where we have considered differential co-expression network in GC and healthy tissues to provide genes that may behave collectively in modules and assess those modules for diagnostic and prognostic performance. Moreover, repurposed drugs that target and reverse the expression of altered context-specific module genes were discovered, which may be used in GC treatment.
In this study, it was studied on gastric tumors and normal gastric tissues, and three datasets obtained from different population groups were integrated. Microarray datasets were statistically analyzed and differentially expressed genes (DEGs) were identified. Mutual DEGs were identified and co-expression network of mutual DEGs was constructed, and altered modules were determined by using tumor and control tissues. The diagnostic and prognostic performance of these modules were evaluated. Furthermore, potential therapeutic drugs that may target and reverse the expression of altered module genes were predicted by using L1000CDS2 signature search engine and resultant drugs or compounds were investigated in literature by text mining method. The altered correlation subnetworks and repurposed drugs presented in this study will be a crucial resource for the development of new disease treatment strategies.
Materials and Methods
Gene expression datasets
The raw data of gene expression datasets of GC from a total of three studies [with Accession No: GSE19826 (Wang et al., 2012), GSE54129 (Liu et al., 2014), and GSE79973 (He et al., 2016)] have been obtained from Gene Expression Omnibus (Barrett et al., 2013) and analyzed to characterize gene expression profiles of GC. The samples in these datasets were taken from the Affymetrix gene chip microarray platform (Fig. 1A). A total of 179 samples were explored, including 133 GC and 46 normal gastric tissue samples.
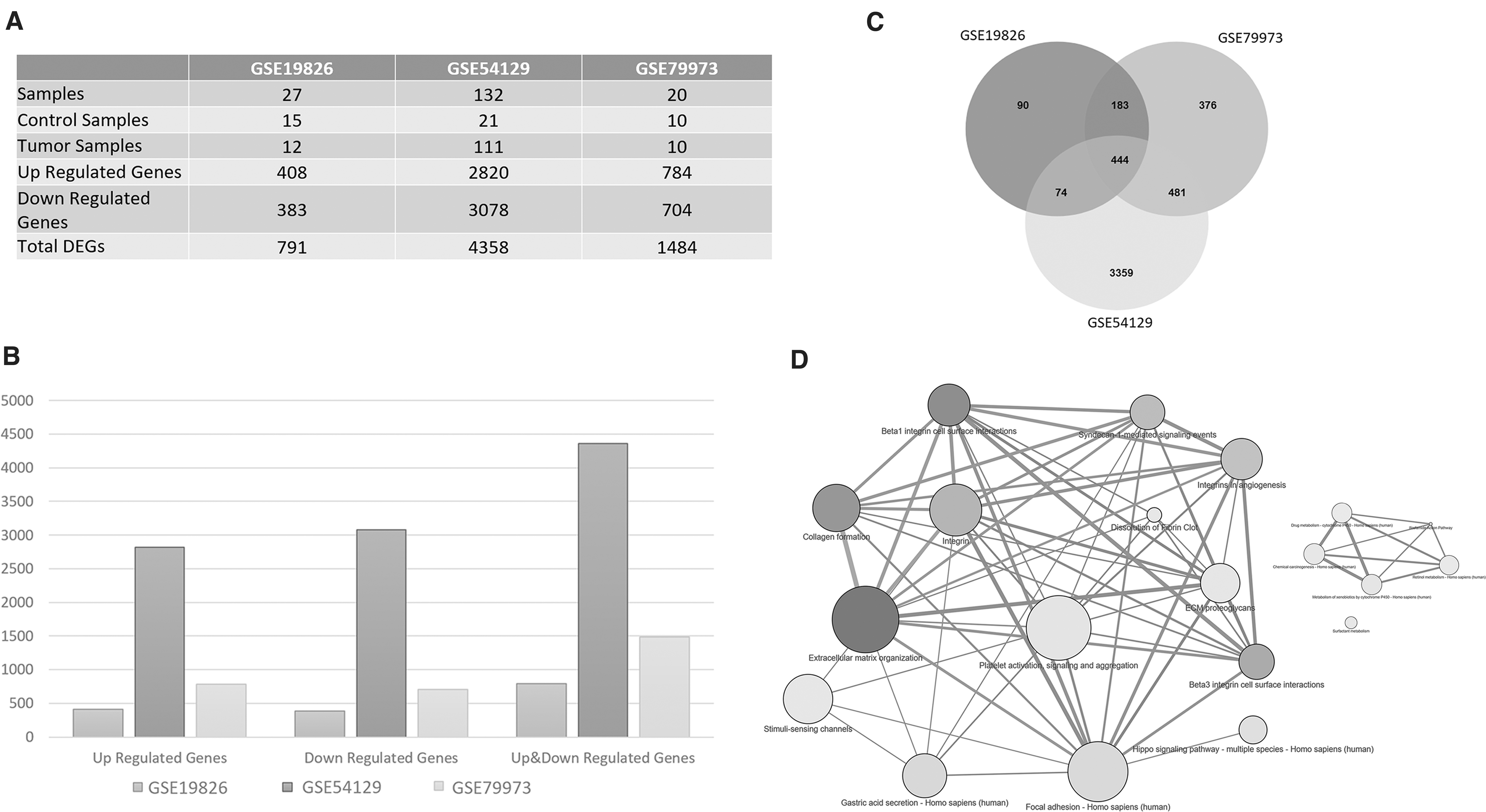
DEGs in gastric cancer datasets.
Independent data, stomach adenocarcinoma (STAD) and stomach and esophagus adenocarcinoma (STES), obtained from The Cancer Genome Atlas (TCGA), which include clinical information about 792 patients, were used in survival analyses. GSE66229 (Oh et al., 2018), which includes gene expression of 400 patients with GC, and normal gastric samples were used for diagnostic analyses.
DEGs and pathways
To identify DEGs, first, each dataset was normalized through the robust multiarray average expression measure (Bolstad et al., 2003), as implemented in the “affy” package (Gautier et al., 2004) in R language (Gentleman et al., 2004). DEGs in each dataset were identified from the normalized log-expression values using the multiple testing options of LIMMA (linear models for microarray data) (Smyth et al., 2005). Benjamini-Hochberg's method was used to control the false discovery rate. A p-value threshold of 0.05 with a fold-change cutoff of 2 was used to determine the statistical significance of DEGs.
Pathway-based overrepresentation analyses of mutual DEGs in all datasets were performed through Metascape (Zhou et al., 2019). Metascape is a web utility that aids to detect significant gene ontology, pathway, and transcription factor (TFs) data. The enrichment analysis was done where the hypergeometric test was used and the significant terms with p-value <0.01 were considered significant. The Bonferroni correction was applied for p-value adjustment.
Co-expression network reconstruction in tumor and normal samples
Expression profiles of mutual DEGs of three datasets were separated as tumor and control samples and two new data lists (consisting of 133 tumors and 46 control samples) were formed using the expression profiles of mutual DEGs. An integrative differential co-expression network analysis methodology (Gov and Arga, 2017) was applied to GC transcriptome datasets to identify differentially co-expressed networks in diseased samples. The mean level of expression of each DEGs was calculated and Z score normalization was applied. It was determined that normalized values are not normally distributed. For this reason, Spearman correlation coefficients (SCC) of mean expression values of mutual DEGs were calculated in diseased and healthy tissues, separately. An SCC cutoff was determined (p < 0.05) and the statistically significant pair-wise correlations of mutual DEGs were identified. It was established a co-expression network in tumor samples.
Identification of altered modules
To identify modules of the co-expression network, The MCODE plugin (Saito et al., 2012) of the Cytoscape (Cline et al., 2007) was used. Modules with at least 10 nodes (genes) and network density ≥0.40 were considered for further analyses. Modules with significantly altered topological metrics (i.e., network density) were considered altered modules compared by SCC values of tumor and control samples. Two threshold conditions (using parameter Ɛ) were defined to classify genes and express the significance of altered expression profiles between cancer and control states:
Genes that show significantly high co-expression in the tumor samples, but no significant co-expression in the control samples. Genes show significantly high co-expression in both tumor and control samples, but change the co-expression direction in tumor and control samples:
Diagnostic and prognostic performance analysis
An independent dataset, GSE66229, was used to identify their potential to differentiate between tumor and control tissues. Raw gene expression values of putative gene groups were extracted and heat maps and dendrograms were drawn through CIMminer online tool (https://discover.nci.nih.gov/cimminer/home.do) according to the Euclidean distance of the biomarker candidates' gene expression.
The prognostic performances of the identified module genes were evaluated using independent GC datasets from TCGA. Cox proportional hazards regression analysis was performed through SurvExpress validation tool (Aguirre-Gamboa et al., 2013). In SurvExpress, the GC adenomocarcinoma (STAD) and STES were grouped into low- and high-risk groups according to their prognostic index. The prognostic capabilities of the resultant genes were identified through Kaplan-Meier (KM) plots and the log-rank test.
Drug repurposing analysis
L1000CDS2 (Duan et al., 2016) is a search engine that provides a listing of small molecules that are represented to mimic or reverse the upregulated and downregulated gene expression profiles. We performed L1000CDS2 analyses using the module gene lists as an input, to find drug or small molecules that reverses the upregulated and downregulated module genes in GC (https://maayanlab.cloud/L1000CDS2/#/index).
Text mining analysis
In this process, repurposed drug candidates were checked in the literature with text mining techniques. All candidates on each module were searched separately on PubMed with different keywords; the published number of articles was stored. “(candidate drug),” “(candidate drug+cancer),” “(candidate drug+gastric cancer)” keywords were scanned in the titles and abstracts of the articles automatically using Python language, and results were parsed with the BioPython package (Cock et al., 2009). Resultant repurposed drugs that are associated with GC and cancer in the literature were sorted by the ratio of the number of articles. Ratios were represented by percentages (i.e, the number of associated “GC and candidate drug” articles/number of total candidate drug-associated articles*100).
Results
Differential gene expression and biological interpretation
We have analyzed three microarray gene expression datasets in this study to identify DEGs in GC compared to controls. We obtained 791 DEGs (FDR <0.05 and logFC = 2) where 408 upregulated and 383 downregulated genes from GSE19826 datasets. Analysis of GSE54129 showed 4358 DEGs (FDR <0.05 and logFC = 2), 2820 upregulated and 3078 downregulated DEGs. Our analysis of GSE79973 showed 1484 DEGs (FDR <0.05 and logFC = 2), 784 upregulated and 704 downregulated genes (Fig. 1A, B). We then sought to detect DEGs that were common among the three datasets to identify the most consistent DEGs, which are likely to be observed in GC samples. Our comparative analysis showed 444 DEGs, which were common among the three above-mentioned GC datasets, which were considered for further downstream analysis (Fig. 1C).
Molecular pathways enriched by the DEGs provide functional association with different gene ontologies and molecular pathways. We performed functional overrepresentation analysis of the common DEGs and detected significant pathways enriched by the DEGs (Fig. 1D). Our analysis showed several key significant pathways enriched by the DEGs. The extracellular matrix (ECM) organization, collagen formation, integrin and proteoglycan related biological processes and pathways, drug metabolism were identified as enriched terms. Moreover, according to Pattern Gene Database (Pan et al., 2013) analysis results, pattern genes of common DEGs were determined as stomach tissue-specific and adipocytes as cell-specific conditions.
Altered co-expressed modules for GC
Identification of key gene modules from the DEGs provides an in-depth understanding of the genes, which behave similar in expression. Thus, we aimed to determine co-expressed key gene modules, using Spearman's gene correlation coefficient. Our analysis determined two crucial gene modules (Fig. 2A). Our enrichment analysis suggested “NABA core matrisome,” “blood vessel development,” “vascular smooth muscle contraction,” “regulation of neuron differentiation,” “NABA ECM glycoproteins,” regulation of peptidase activities, and “ossification” were enriched by module 1 genes (Fig. 2B). However, “PID Integrin 3 pathways,” “regulation of transmembrane receptor protein serine/threonine kinase signaling pathway,” and “post-translational protein modification” pathways were enriched by module 2 (not by modules 1).
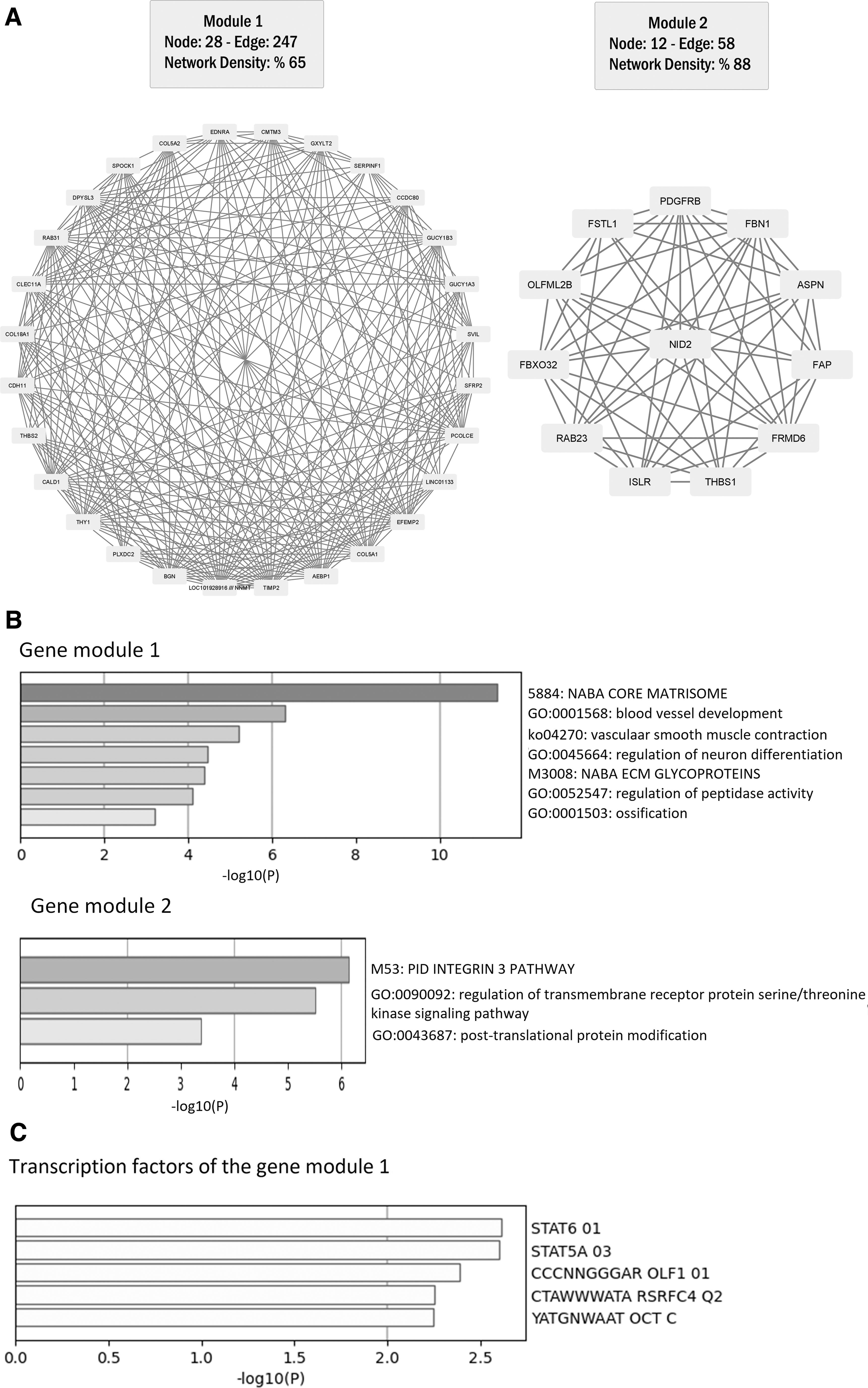
The co-expressed gene modules and their biological importance in gastric cancer.
The genes are regulated at transcriptional and post-transcriptional levels. To detect the TFs that may regulate the transcription of DEGs, we performed an analysis that showed STAT6 (signal transducer and activator of transcription 6) and STAT5A (signal transducer and activator of transcription 5A), among others that may regulate the DEGs of module 1 (Fig. 2C). We have not found any enriched significant TFs for module 2.
Altered co-expressed modules as potential diagnostic and prognostic gene signatures
Using an independent data set, the expressions of the module genes were plotted as dendrogram by using Euclidean distance (Fig. 3). GC tumor and control samples were divided into almost two main groups, and it was determined that modules could differentiate between tumor and normal gastric tissues.

Clustering analysis of co-expressed gene modules on an independent dataset by using Euclidean distance. Black and gray lines indicate tumor and normal samples, respectively.
To predict the survival of GC using the co-expressed modules, we performed KM plot and long-rank hazards text using independent RNA-Seq data from the TCGA database. We have shown that the dysregulated expression of two gene co-expressed modules was associated with the worst outcome in both STAD and STES (Fig. 4). Our analysis suggested the identified modules may be used as prognostic biomarkers in GC.

Prognostic performance analysis of co-expressed gene modules with Kaplan-Meier plot and long-rank hazards test. Black and gray lines represent high and low risks, respectively.
Candidate repurposed drugs and small molecules for GC
Considering the module genes as prognostic targets in GC, we have used the LINCS L1000 characteristic gene expression direction search engine. The list of module genes was entered to L1000CDS2 web tool to search for agents that can reverse the module's gene expression. Fifteen drugs or small molecules were identified as repurposed drugs, showing potential with an overlap score of >0.3 to reverse expression profiles on upregulated and/or downregulated module genes in cell lines (Table 1). Repurposed drug candidates were examined in the literature with text mining techniques. According to results, the repurposed drugs that were studied in GC and cancer were determined (Fig. 5).

PubMed results and order of candidate drugs and small molecules by text mining method and sorting by the ratio of the number of articles.
Repurposed Small Molecules Showing the Reversal Gene Expression Profiles in Several Cell Lines, for Gastric Cancer
Repurposed drugs or small molecules, including trichostatin A (TSA), albendazole, brefeldin A, and vorinostat, which are known drug candidates for GC treatment were found. Especially, belinostat and danusertib have been studied in many cancer-related studies (83.45% and 76.67%, respectively), and both of them are not investigated in GC-associated research, except for two articles for danusertib. Therefore, these two drugs show potential for use in the treatment of GC. Furthermore, kinetin riboside and withaferin-a have limited cancer-related studies (46.15% and 51.39%, respectively) and both of them are not investigated in GC-associated research.
Discussion
GC is one of the prevalent cancers affecting many people worldwide. Despite many efforts in GC research, still, the biomarkers and effective therapeutic agents are not yet clearly found. Our proposed systems biology method to identify repurposing potential drugs was applied with the following: (i) three publicly accessible datasets were ostensibly used for this study. (ii) The analyses have identified altered gene expression signatures, from a GC diagnostics standpoint. (iii) These findings were subsequently examined for their value for the prognosis of GC as well as being used as guideposts for drug repurposing analysis.
We investigated comprehensively the microarray gene expression datasets and showed the 444 mutually dysregulated DEGs in three GC datasets. Our gene expression analysis showed the presence of two key gene modules that have diagnostic and prognostic capabilities. Investigating the molecular pathways is critical to clarify the potential mechanism of GC; thus, we also performed gene enrichment analysis. It showed the significant enrichment of ECM, ECM regulators, and NABA core matrisome, which are in agreement with previous findings in GC and other cancer (Abbaszadegan et al., 2020; Moreira et al., 2020; Yuzhalin et al., 2018). Our study suggests enrichment of PID Integrin 1 pathway in GC.
The integrins are key mediators that connect cancer cells with ECM (Aoudjit and Vuori, 2012; Shin et al., 2012). Our study suggests enrichment of PID Integrin 1 pathway in GC. The integrins are key mediators that connect cancer cells with ECM (Aoudjit and Vuori, 2012; Shin et al., 2012).
Moreover, it was investigated that the TFs may regulate DEGs and co-expressed gene modules. Among the TFs, STAT6 recently showed the reduction of cell proliferation, migration, or invasion of GC cells upon inhibition of STAT6/Anoctamin-1, suggesting crucial role of STAT6 in GC (Lu et al., 2018).
According to drug repurposing and text mining analysis, the top 3 highest scores were investigated in the literature. The first two, TSA and belinostat (S1085), share the same score as 0.4348. The inhibitory properties of TSA as histone deacetyltransferase (HDAC) in GC has been shown by in vitro studies (Li et al., 2016; Sun et al., 2015). The mechanism underlying this inhibitory effect is still unknown; however, the changes in propanoate metabolism pathway after 330 nM TSA (100 ng/mL) application for 24 h in GC epithelial cell line, BGC-823, were suggested in a study (Li et al., 2017).
In another study, decreased expression of glycoprotein nonmetastatic melanoma protein B (GPNMB) in BGC-823 cell line was shown after 75 ng/mL TSA treatment for 48h (Ruan et al., 2014, p. 201). Although there are some studies that investigate TSA-GC cell line interaction, there is a still a very big gap for studies including clinical samples. Belinostat (score: 0.4348) and vorinostat (score: 0.3913) are Food and Drug Administration (FDA)-approved histone deacetylase inhibitors (HDACi) (Singh et al., 2018). Belinostat is indicated for the treatment of patients with relapsed or refractory peripheral T cell lymphoma (Belinostat: Uses, Interactions, Mechanism of Action, 2021).
The results of phase I pharmacokinetic study of belinostat in cancer patients having different tumor types (1 GC patient over 72) indicated that there was no significant increase in liver toxicity in patients having liver dysfunction (Takebe et al., 2019). Neither in vitro nor in vivo or clinical studies investigating the effect of belinostat on a GC cell line or patients was found in the literature. This made it important to study the belinostat drug molecule in GC disease.
Vorinostat (score: 0.3913) was approved by FDA in 2006 and has been used in the treatment of advanced primary cutaneous T cell lymphoma (Mann et al., 2007). In a study, reactivated RUNX3 expression in GC cell lines after vorinostat application triggered cell apoptosis (Huang et al., 2007). In addition, the vorinostat drug molecule was suggested as a strong candidate or a part of combined drug therapy for GC treatment as a result of gene signature analysis (Claerhout et al., 2011). However, in phase II clinical study, metastatic or unresectable GC patients were treated with the combination of vorinostat, capecitabine, and cisplatin.
The results showed that this drug combination was not better when compared to the standard therapy in GC patients (Yoo et al., 2016). In our study, vorinostat is calculated as the second and third most related candidate drug molecule, both by L1000CDS2 tool and text mining method, respectively.
Albendazole (score: 0.3478) is an anthelmintic drug, an antiparasitic drug. It has been used in animals and humans since the 1980s and has low toxicity. It disrupts microtubule assembly and glucose uptake in parasites, so their proliferation is inhibited (Zhang et al., 2017). Besides its anthelmintic properties, its effects on different cancer types have been investigated in drug repositioning studies (Chai et al., 2021). In a study, three different GC cell lines were studied in the presence of 0.01–1.5 μM albendazole in cell culture. Cellular apoptosis as a result of microtubule disruption was shown in dose-dependent manner (Zhang et al., 2017).
In another study, albendazole was applied to different types of GC cell lines between 10 and 100 μM concentrations. The researchers concluded that ABZ may affect some oncogenic TFs such as STAT3/5 and displays antitumoral activity (Yang et al., 2021). In our study, STAT6 is also suggested as a key regulator for the selected gene module, so the relationship between repurposed small molecules and STAT6 expression in GC cell lines may play an important role in cancer prognosis.
In one study, TSA application enhanced cisplatin-induced ototoxicity in rat cochlear hair cell culture by the inhibition of STAT6 and IL-4 expression (Hsu et al., 2015). The effects of belinostat on nine different lung squamous cell carcinoma cell lines were investigated. There was a small decrease in the expression of STAT6 and a significantly higher expression of STAT3 in Calu-1 and H520 cell lines (Kong et al., 2017). The interaction between vorinostat drug molecule and STAT6 has been studied in different types of cancers. The inhibition of STAT6 phosphorylation by vorinostat application and increase in cellular apoptosis was shown in Hodgkin lymphoma cell lines (Buglio et al., 2007).
In addition, decreased expression of STAT6 in cutaneous T cell lymphoma cell lines was revealed in cell culture studies in which vorinostat was applied in apoptotic concentrations (Duvic and Zhang, 2006). In short, several repurposed drug candidates, including TSA, vorinostat, parthenolide, panobinostat, brefeldin A, belinostat, and danusertib, which may be used in GC treatment, were identified. Belinostat and danusertib are determined as novel drug candidates for GC treatments because they have been studied in many cancer-related studies, although there are no GC-related studies, including these drugs.
Although our study provides critical genes, TFs, pathways in GC, and repurposed drugs or small molecules for GC treatment, several limitations of the study should be noted as we could not validate the repurposed drug effect on the GC cells in a wet-lab setting; thus, text mining analysis and interpretations regarding candidate drugs should be taken cautiously into consideration. While our work has demonstrated the benefit and usefulness of gene expression analysis of GC in bringing out the novel drugs and small molecules that can may be potential therapeutic targets in GC, the need for improved analysis and prospective clinical studies trials is still imperative.
Conclusions
This study aimed to identify altered gene expression signatures, from a GC diagnostics standpoint, and module genes were subsequently examined for their value for prognosis of GC as well as being used as guideposts for drug repurposing analysis. We detected two co-expressed gene modules could differentiate between tumor and normal gastric tissues. Furthermore, these genes were significant prognostically in predicting the survival outcome in GC in independent datasets. Several repurposed drug candidates, including TSA, vorinostat, parthenolide, panobinostat, brefeldin A, belinostat, and danusertib, which may be used in GC treatment, were identified.
In this study, the identified co-expressed module genes as prognostic and diagnostic biomolecules, Belinostat and Danusertib as novel drugs, STAT6 as a TF may facilitate critical research directions and development of new strategies for GC treatment and prognosis. Considering the immense importance of the identified gene signature, pathways, and repurposed drugs, we now propose experimental validations of these in a wet-lab experimental setup to establish them in clinical use.
Footnotes
Acknowledgments
The scholarships under the TÜSEB project 2019-TA-01/3436 provided to TYD are greatly acknowledged.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This study is supported by Health Institutes of Turkey (TÜSEB) in the context of the project 2019-TA-01/3436.