
Abstract
Planetary agriculture stands to benefit immensely from phytopathogen diagnostics, which would enable early detection of pathogens with harmful effects on crops. For example, Phytophthora palmivora is one of the most destructive phytopathogens affecting many economically important tropical crops such as coconut. P. palmivora causes diseases in over 200 host plants, and notably, the bud rot disease in coconut and oil palm, which is often lethal because it is usually detected at advanced stages of infection. Limited availability of large-scale omics datasets for P. palmivora is an important barrier for progress toward phytopathogen diagnostics. We report here the mycelial proteome of P. palmivora using high-resolution mass spectrometry analysis. We identified 8073 proteins in the mycelium. Gene Ontology-based functional classification of detected proteins revealed 4884, 4981, and 3044 proteins, respectively, with roles in biological processes, molecular functions, and cellular components. Proteins such as P-loop, NTPase, and WD40 domains with key roles in signal transduction pathways were identified. KEGG pathway analysis annotated 2467 proteins to various signaling pathways, such as phosphatidylinositol, Ca2+, and mitogen-activated protein kinase, and autophagy and cell cycle. These molecular substrates might possess vital roles in filamentous growth, sporangia formation, degradation of damaged cellular content, and recycling of nutrients in P. palmivora. This large-scale proteomics data and analyses pave the way for new insights on biology, genome annotation, and vegetative growth of the important plant pathogen P. palmivora. They also can help accelerate research on future phytopathogen diagnostics and preventive interventions.
Introduction
Phytophthora palmivora is a highly destructive phytopathogenic hemibiotrophic oomycete that causes diseases in an extensive range of over 200 host plants, most of which possess economic significance (Perrine-Walker, 2020). Some of the important diseases caused by P. palmivora in the tropics include the bud rot of coconut and oil palm; black pod, stem canker and chupon wilt in cocoa; root rot in durian; fruit rot in papaya; crown and root rot in citrus and leaf fall in rubber.
Numerous genome sequencing initiatives have been undertaken in Phytophthora spp. (Gao et al., 2015b). These efforts have unearthed the vast repertoire of the pathogenicity factors and effectors (apoplastic and translocated), which possess key roles in promoting the infection process (Chepsergon et al., 2021; Wang and Jiao, 2019). In addition, these studies have revealed the expansion of gene families encoding hydrolytic enzymes, including carbohydrate-active enzymes (CAZymes) and proteases, which aid in the breakdown of integrity of plant cell walls and boost the pathogenesis process (McGowan, 2020).
Sequencing an organism's genome can provide details about the entire set of genes in that organism, which are usually predicted using gene prediction algorithms. Protein-coding genes are often identified from the genome sequence based on ab initio prediction and homology-based prediction, ably assisted by transcript evidence (Keilwagen et al., 2018). Despite the immense progress in developing gene-prediction algorithms, protein expression-based validation by a global proteomic analysis study provides protein-level evidence for genome annotation.
A mass spectrometry (MS)-based global proteomics analysis is used to identify proteins and their post-translational modifications (PTMs) in a biological sample and also to determine the expression pattern or abundance of a protein and/or PTMs (Deracinois et al., 2013). In general, a global proteomic analysis for genome annotation validation is hypothesis free and offers a useful array of sequences validated by mass spectra (Deracinois et al., 2013). Technological progress achieved in MS-based global proteomics can now provide multi-dimensional insights on oomycete biology, and equally important, reveal the possible metabolic targets for control of oomycetes. Given the enormous economic threat posed by P. palmivora, the bud rot pathogen, on coconut plantations in the tropics, this study makes a contribution with a global proteomic analysis to identify the proteins in mycelia of P. palmivora based on the liquid chromatography-MS/MS technology.
Materials and Methods
Isolation and in vitro culturing of P. palmivora
A highly virulent isolate of P. palmivora, TR-PP-2, isolated previously in our laboratory (Gangaraj and Rajesh, 2020; Gangaraj et al., 2021), was used for this study. The isolate was maintained in carrot agar medium in Petri plates, kept in a growth chamber with a constant temperature (25°C ± 2°C) and humidity (95%). An individual isolate was inoculated onto the carrot broth medium and maintained at constant temperature (25°C ± 2°C) for the formation of mycelial mats. Seven-day-old mycelial mats were filtered, washed thoroughly with sterile distilled water, and then dried using sterile filter papers. Permission to undertake this research has been obtained from the Institute Research Committee (IRC) meeting of ICAR-CPCRI held from June 22–25, 2021 (ICAR-CPCRI Project No. 1000761030).
Extraction of proteins, digestion, and fractionation from mycelial mats of P. palmivora
Protein extraction from the mycelial mats was carried out following the procedure of Krishnaswamy et al. (2019) with minor modifications. In brief, the fungal mycelial mat was cut into small pieces and extracted in 8 M urea buffer with 1 mM EDTA disodium salt, 40 mM Tris-HCl (pH 6.8), and 4% (v/v) SDS using a Bead Beater (Fast Pre-24) for 10 min in cold condition. The extract was centrifuged for 12 min at 12,000 g and 4°C to collect the supernatant. Later on, the supernatant was precipitated in chilled acetone (supernatant: acetone ratio of 1:9) by overnight incubation at −20°C. After centrifugation, the protein pellet was reconstituted in 50 mM TEABC (guanidine hydrochloride will be in the supernatant phase).
Protein estimation was performed using bicinchoninic acid assays (cat. no. 23250; Thermo Scientific Pierce) using a micro-volume spectrophotometer (Thermo Scientific Multiskan Sky, 1530) and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) to normalize the protein amounts to perform in-solution digestion. The protein (500 μg) dissolved in 50 mM TEABC buffer was used for digestion following Mohanty et al. (2020) with minor modifications. In brief, 100 mM dithiothreitol, a reducing agent, was added to the digested protein, and the contents were incubated at 60°C for 20 min. Thereafter, 100 mM of freshly prepared iodoacetamide, the alkylating reagent, was added at a final concentration of 20 mM and incubated for 10 min. Promega trypsin at a ratio of 1:20 (enzyme:protein ratio) was used for digestion and incubated at 37°C overnight.
Acidification of the solution was performed with the addition of 0.1% formic acid to quench the trypsin activity, and the protein digestion was confirmed using SDS-PAGE. The peptides were further desalted using C18 Stage Tips and then fractionated to six fractions using the Strong Cation Exchange method to remove salt and other contaminations. The column was washed with 100% acetonitrile, then equilibrated using 50 μL of 0.2% trifluoroacetic acid (TFA) twice. The sample was loaded and washed twice using 50 μL of 2% TFA; the peptides were eluted in 50 μL of elution buffers containing different concentrations of ammonium acetate (50–300 mM). The eluents were dried using SpeedVac (Thermo Scientific-RC110B). A schematic representation of the extraction of proteins and proteomic analysis is given in Figure 1.

Schematic representation showing the proteomic analysis of Phytophthora palmivora. The protein identification was performed using MASCOT and SEQUEST HT search engines and the unassigned spectra from each search were used in the subsequent search.
MS analysis
The fractionated and dried peptides were cleaned using the C18 StageTip-based method. The C18StageTip cleanup column was charged with 100% acetonitrile (ACN), followed by 0.1% formic acid for column equilibration, then sample loading and finally elution was carried out with 40% acetonitrile in 0.1% formic acid. An Orbitrap Fusion Tribrid mass spectrometer (Thermo Fischer Scientific, Bremen, Germany) connected to Easy-nLC-1200 nanoflow liquid chromatography system (Thermo Scientific) was used for data acquisition. The peptides were reconstituted in 0.1% formic acid and loaded onto a 2 cm trap column (nanoViper, 3 μm C18 Aq) (Thermo Fisher Scientific). Peptides were separated using a 15 cm analytical column (nanoViper, 75 μm silica capillary, 2 μm C18 Aq) at a flow rate of 300 nL/min. The mobile phase solvents (80% acetonitrile in 0.1% formic acid) were set as gradients of 5–35% for 110 min.
The total run time for each fraction was 140 min. Global MS survey scan was carried out at a scan range of 400–1600 m/z mass range (120,000 mass resolution at 200 m/z) in a data-dependent mode using an Orbitrap mass analyzer. The maximum injection time was 5 msec. Only peptides with charge states of 2–6 were considered for analysis, and the dynamic exclusion rate was set to 30 sec. For MS/MS analysis, data were acquired at top speed mode with a cycle time of 3 sec and subjected to higher collision energy dissociation with 34% normalized collision energy. MS/MS scans were carried out at a range of 100–1600 m/z using an Orbitrap mass analyzer at a resolution of 60,000 at 200 m/z. The maximum injection time was 120 msec.
Analysis of MS/MS and proteomic data
The raw data obtained from the Orbitrap MS/MS was analyzed using the Proteome Discoverer (PD) platform, version 2.2 (Thermo Scientific). The MS/MS data were searched against the P. palmivora database and common contaminants using the PD platform integrated with Sequest HT and Mascot (Version 2.4) search algorithms. Furthermore, the unassigned spectra were also searched sequentially against the protein databases of P. infestans, P. sojae, P. parasitica, P. meadii, and P. megakarya.
These protein databases were obtained from Refseq, NCBI and the number of proteins in each database is given in Table 1. Protein sequences were theoretically digested with trypsin set as protease enzyme with a maximum of two missed cleavages and, minimum peptide length with seven peptides. The PD search parameters were set with precursor mass tolerance of 20 ppm, fragment mass tolerance of 0.05 Da, and 1% false discovery rate at peptide spectrum match (PSM), peptide, and protein levels. Oxidation of methionine and protein N-terminal acetylation were set as variable modification and carbamidomethylation of cysteine as fixed modification.
Details on Postrun Proteomic Analysis of Phytophthora Palmivora Mass Spectrometry Spectra
PSM, peptide spectral match.
Quality check of peptides from unassigned spectra
Retention time (RT) prediction of peptides, identified in the database searches of unassigned spectra, was carried out using AutoRT. An ensemble base model was created using PSMs with RT information from P. palmivora database search (First search). Ensemble RT model was trained with the help of a small dataset from a global proteome experiment. Model training was performed with 40 epochs and a batch size of 64. Maximum peptide length was set to 51 and amino acids with modifications such as carbamidomethylation (C), oxidation (M), and phosphorylation (STY) were defined with numbers 1, 2, and 5, respectively, in the peptide sequence.
Finally, the RT prediction for the PSMs identified from database searches of P. infestans, P. sojae, P. parasitica, P. meadii, and P. megakarya were performed against the trained ensemble RT model. Difference between the experimental and predicted RT was used to calculate the absolute RT error (min) and median absolute RT error (MAE) for PSMs from each search.
Functional analysis of proteins
Functional analysis of all the proteins identified was categorized based on their biological processes (BP), cellular components (CC), and molecular functions using the BLAST2GO tool (https://www.blast2go.com). The domain analysis of all the proteins was undertaken using Web-CD Search Tool (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi) to identify the major protein superfamilies to which they belong. Pathway analysis of the proteins was carried out using the KEGG pathway database (https://www.genome.jp/kegg/pathway.html). The data obtained were compared with published articles on the proteins and signaling in Phytophthora spp. and Saccharomyces cerevisiae (yeast).
Data availability
The MS raw data and result files were deposited to the ProteomeXchange Consortium using the PRIDE (PRoteomicsIDEntifications database) partner repository (https://www.ebi.acuk/pride/markdownpage/submitdatapage) with the dataset identifier PXD028988.
Results and Discussion
The limited availability of large-scale omics datasets for P. palmivora is an important barrier for early diagnosis and intervention to fight this phytopathogen. The proteomics data generated through this study contributes to addressing this gap.
Proteomic analysis of a virulent P. palmivora isolate, obtained from mycelial mat grown for seven days in carrot broth, was carried out using an Orbitrap Fusion Tribrid mass spectrometer. The postrun analysis of the generated MS/MS spectra was carried out using Sequest HT and Mascot search engines against selected protein databases of Phytophthora spp. The number of proteins in each database, the total number of identified proteins, peptides, PSMs, and unassigned spectra for each search are given in Table 1. A complete list of identified proteins and their accession numbers, protein definition, unique peptide sequences, and PSMs for each search are given in Supplementary Tables S1–S6.
In this study, performing the postrun analysis of LC-MS/MS data against the P. palmivora database resulted in the detection of 4182 proteins. Many spectra that were not assigned to any other proteins in the database were considered “Unassigned spectra.” To maximize the identification of proteins, the unassigned spectra were subjected to proteogenomic protocols by searching against a set of related species of Phytophthora such as P. infestans, P. sojae, P. parasitica, P. meadii, and P. megakarya in a sequential manner (Table 1). This proteogenomic approach resulted in the detection of additional proteins, thus giving a total of 8073 proteins (Table 1).
Phytopathogenic oomycetes rely on a multitude of metabolic pathways to undertake transition between different developmental stages and host colonization and subsequent infection. The characterization of proteins, expressed specifically at various developmental stages, can divulge information on life stage-specific essential proteins. Therefore, an in-depth proteomic study of oomycetes can complement the genome or transcriptome strategies in enriching the knowledge on key determinants of dynamics of growth, reproduction, survival, virulence, and other cellular processes needed for colonization of plants and their interactions.
Isobaric tags for relative and absolute quantitation (iTRAQ) method has been utilized to interrogate the alterations in the protein expression patterns in Phytophthora capsici on exposure to three novel fungicides, pyrimorph (Pang et al., 2015), SYP-14288 (Cai et al., 2019), and cinnamaldehyde (Wang et al., 2021); the metabolic pathways targeted by these fungicides have also been revealed. A similar study, using tandem mass tag (TMT) labeling, has revealed the perturbation of fatty acid metabolism by propyl gallate, which resulted in the inhibition of growth of P. sojae (Liu et al., 2020).
Quantitative proteomics study of asexual (mycelium and cyst) stage of P. capsici (Pang et al., 2015) and sexual (oospore) and asexual (mycelium and cyst) stages of P. sojae (Zhang et al., 2020), undertaken using iTRAQ and TMT labeling, respectively, revealed underpinnings of the biology of these oomycetes, specifically proteins that are upregulated at each developmental stage. In addition, this study also put forth potential targets for controlling these phytopathogens. A comparative MS-based mycelial and extracellular proteome analysis of three Phytophthora species, P. chlamydospora, P. gonapodyides, and P. pseudosyringae, has revealed numerous proteins with potential roles in pathogenicity and osmotrophy and provided insights into the behavior and lifestyle of these three species (McGowan, 2020).
In any newly annotated genome, computational strategies are utilized to assemble the genomic sequence and prediction of protein-coding genes. Errors can creep in because of multiple factors, including quality of genome sequence reads, assembly issues, nature of reference assembly used, missing genomic regions, and so on. As a result, the predicted proteome database may contain errors in the sequence of proteins and possibly also miss many bona fide proteins. Comparative genomic analysis often accommodates sequences of closely related species to add to the discovery of protein-coding regions.
A global proteomic analysis provides invaluable data to validate these computationally predicted tools. However, a search of MS data can fetch mass spectra evidence for only those peptides that are represented in the predicted protein database. All other MS/MS spectra remain unassigned because of an incomplete match. The MS search algorithms will identify additional peptides from the unassigned MS/MS data if we search against other hypothetical databases of closely related species. We gained identification of a large number of additional proteins from an iterative search of unassigned mass spectra against protein databases of P. infestans, P. sojae, P. parasitica, P. meadii, and P. megakarya.
Quality check of peptides
A proteome database search of unassigned spectra cannot distinguish true and false identified PSMs. This can either be because of the large size of the proteome database or a low number of input spectra when compared with the database (Verbruggen et al., 2021). Nonetheless, the difference between the predicted peptide RT with its experimentally observed RT can be used as a feature to increase the confidence on PSMs identified from the search of unassigned spectra. Therefore, we carried out machine-learning-based peptide RT prediction for PSMs identified from the database searches of unassigned spectra using an ensemble model using AutoRT (Wen et al., 2020).
The RT prediction of PSMs identified from the database searches of unassigned spectra were very close to the experimental RT values based on the MAE (Supplementary Table S7). The MAE for PSMs identified from the P. infestans (26,590), P. sojae (7952), P. parasitica (2939), P. meadii (3430), and P. megakarya (3,527) were 2.06, 2.17, 2.30, 1.88 and 2.59 min, respectively (Fig. 2A). This shows that the bulk of the peptides identified in the database searches was within ∼2 min of the predicted RT, indicating no or a very low number of false-positive hits.
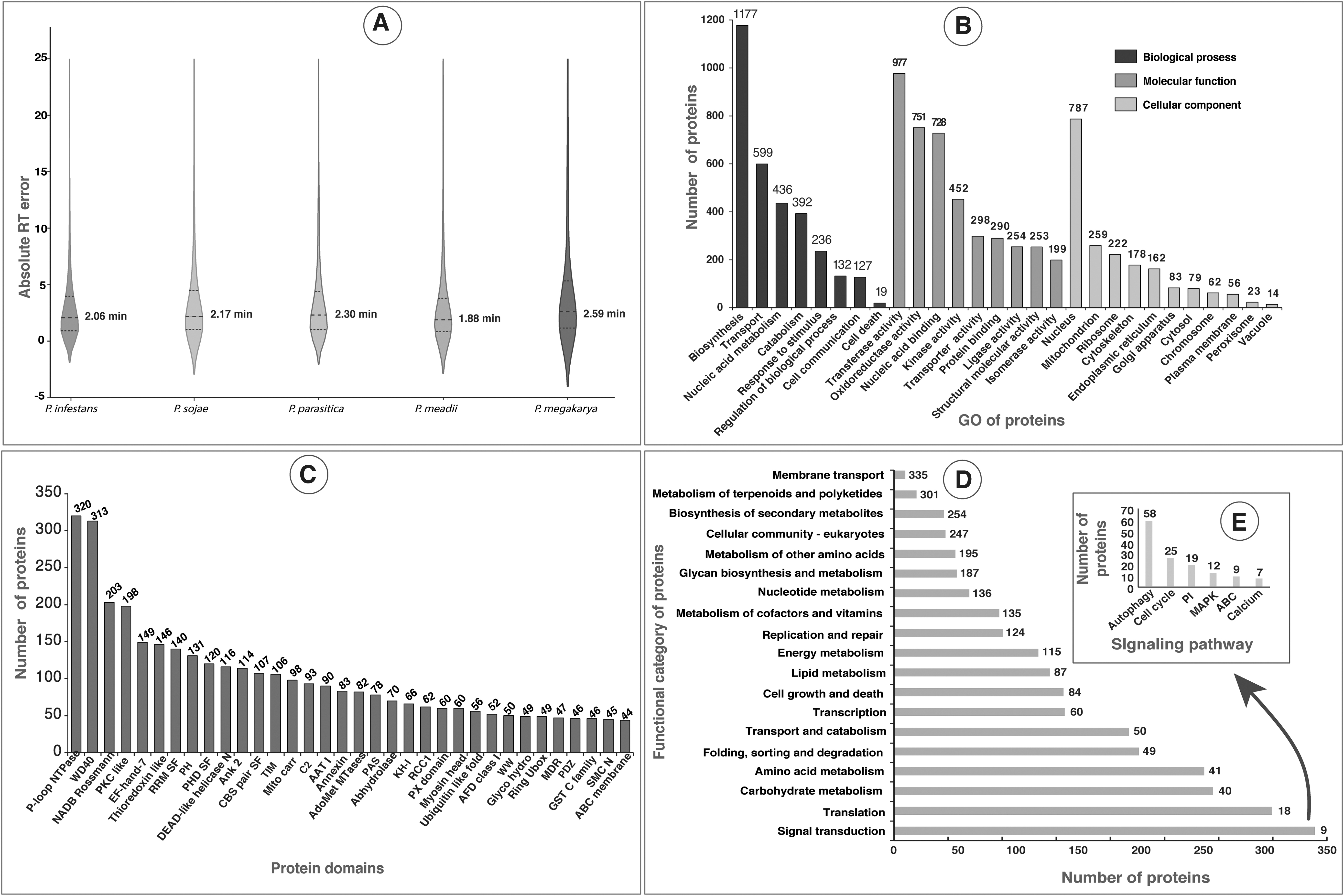
Analysis of P. palmivora proteins based on functions, domains, and KEGG pathway.
Functional analysis of P. palmivora proteins
Blast2GO suite is a comprehensive bioinformatics tool designed for functional annotations of novel sequences and data mining on the resulting annotations based on Gene Ontology (GO) (Conesa and Götz, 2008). In this in silico approach, Blast2GO optimizes function transfer from homologous sequences through an elaborate algorithm that considers similarity, the extension of the homology, the database of choice, the GO hierarchy, and the quality of the original annotations. The GO-based functional classification of P. palmivora proteins using BLAST2GO annotation tool resulted in 4884 (60.5%) proteins mapping to BP, 4981 (61.7%) proteins to molecular functions, and 3044 (37.7%) proteins to different CC (Fig. 2B). The largest group of proteins among BP belonged to “biosynthesis” (1177 proteins) followed by “transport proteins” (559). In addition, we also identified 236 and 127 proteins, which were associated with “response to stimulus” and “cell communications,” respectively.
The detection of many proteins involved in BP such as biosynthesis and transportation could be owing to the rapid growth of P. palmivora on a rich nutrient medium under laboratory conditions. Molecular functional (MF) analysis revealed the majority of the proteins with transferase (977), oxidoreductase (751), nucleic acid binding (728), and kinase (452) activities, supporting the major role of proteins in the growth of fungus. In addition, the GO analysis also revealed the detection of the largest group of nuclear proteins (787) under CC in comparison with all other organellar proteins. Further experimental validation of proteins predicted with specific BP, MF, and CC are necessary to conclude the results.
The domain analysis, undertaken using Web-CD Search Tool, revealed that the majority of the domains belonged to the P-loop NTPase domain superfamily (Walker A motif) followed by the WD40 domain (Fig. 2C). The P-loop NTPases are classified into (1) the KG (kinase-GTPase) class, which includes Ras-like GTPases and its circularly permutated YlqF-like; and (2) the additional strand catalytic E class, which includes ATPase Binding Cassette (ABC), DExD/H-like helicases, 4Fe-4S iron-sulfur cluster binding proteins of NifH family, RecA-like F1-ATPases, and ATPases Associated with a wide variety of Activities (https://www.ncbi.nlm.nih.gov/Structure/cdd/cl09099).
The P-loop NTPases are known to be involved in diverse cellular functions and play key roles as signal-generating entities in both prokaryotes and eukaryotes (Leipe et al., 2004). Similarly, the WD40 domain, which has been reported to exhibit varying functions, in signal transduction, processing of pre-mRNAs, assembly of the cytoskeleton and coordinating protein–protein interactions (Jain and Pandey, 2018), was found to be a part of cyclophilin-encoding genes, which aid host infection in Phytophthora spp. (Gan et al., 2009). However, confirmation of the current results needs further experiments.
Pathway analysis of P. palmivora proteins
Pathway analysis using the KEGG database showed the contribution of 2467 proteins in different signaling pathways. Of these proteins, a maximum number of 335 proteins showed their role in “signal transduction,” followed by “translation” (301 proteins) (Fig. 2D). In addition, the majority of the proteins detected in P. palmivora belonged to the normal cellular pathways, such as cell division, transport, and degradation.
In this study, the proteins and their domains detected in P. palmivora (Supplementary Table S8) possessed roles in different signaling pathways such as autophagy, cell cycle, phosphatidylinositol (PI), mitogen-activated protein kinase (MAPK), cAMP, ABC transporter, and calcium signaling as observed in Phytophthora spp. and yeast (Gustin et al., 1998; Tamaki, 2007; Wera et al., 2001). Probably, these are the signaling pathways essential for the normal growth and development of P. palmivora. A detailed KEGG pathway analysis annotates proteins with their role in filamentous growth, induction of sporangia, and autophagy in P. palmivora, as explained in the section “Regulation of filamentous growth and induction of sporangia.”
Regulation of filamentous growth and induction of sporangia
Filamentous growth in fungal species is mainly regulated by the availability of nutrients (Sengupta et al., 2010) and is critical in the case of pathogenic fungi such as P. palmivora to induce host–pathogen attachment, invasion into the host tissue, and to induce virulence (Brückner and Mösch, 2012; Cullen and Sprague, 2012). Signal transduction pathways such as MAPK, rat sarcoma/protein kinase A (RAS/PKA), sucrose nonfermentable (SNF), and target of rapamycin (TOR) have been well studied in S. cerevisiae in the context of regulation of filamentous growth (Bardwell, 2006; Dohlman and Slessareva, 2006; Saito, 2010). However, no detailed information is available on the signal transduction pathway in phytopathogenic P. palmivora.
Limited information is available on the related pathogenic oomycetes such as P. sojae and Peronophythora litchii, MAPKs have been implicated in the development of oomycetes, release and viability of zoospores, germination of oocysts, cleavage of sporangia, sexual reproduction, tolerance to diverse stresses, pathogenicity, and/or virulence (Gao et al., 2015a; Huang et al., 2021; Jiang et al., 2018; Li et al., 2010). Therefore, the proteins of MAPK-mediated signal transduction pathway and its associated signaling pathways such as PI signaling, calcium (Ca2+) signaling, cell cycle regulation, and TOR-mediated autophagy signaling pathways detected were analyzed and compared with that of S. cerevisiae along with a model figure (Figs. 3 and 4). This will help understand the probable signaling pathways and associated cross-talks that are responsible for the filamentous growth and development in P. palmivora.

Probable signaling pathways in P. palmivora have a role in the regulation of filamentous growth and induction of sporangia. All the proteins annotated in the KEGG pathway with their role in PI signaling, Ca2+ signaling, MAPK signaling, cell cycle, ABC transporter functions are given in the figure. ABC, ATPase binding cassette; MAPK, mitogen-activated protein kinase.
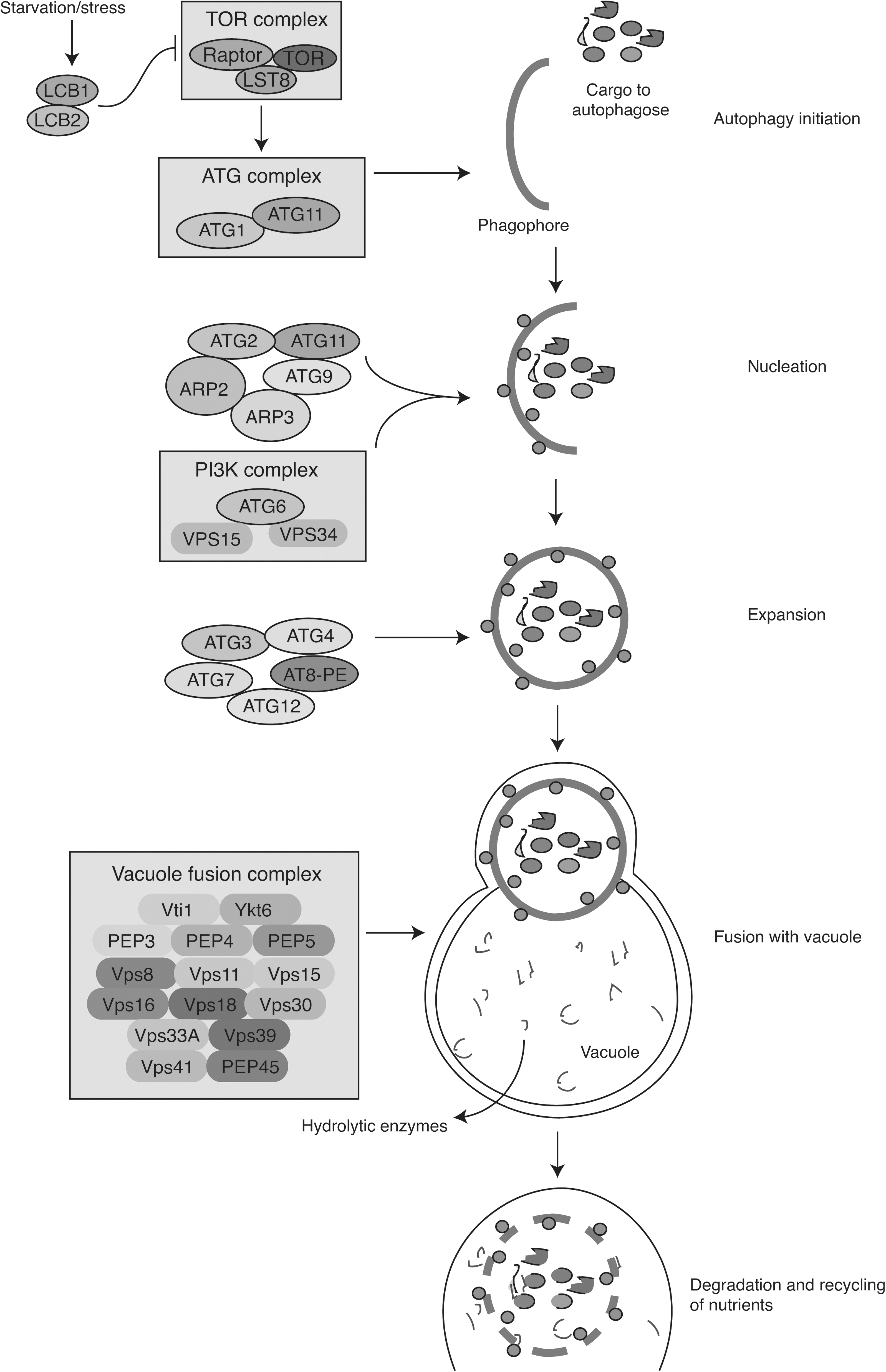
Schematic diagram of autophagy in P. palmivora. Different proteins detected in P. palmivora are shown with their probable role in each stage of autophagy starting from initiation to degradation. The proteins and their role were compared with that of yeast autophagy.
The phosphorylated derivative of PI, located on the plasma membrane, is the starting point for several kinase activities. PI is synthesized from cytidine diphosphate- (CDP-)diacylglycerol and myo-inositol by PI synthase (PIS). Its head group, inositol, can be phosphorylated to PI mono, bis-, and trisphosphates, ultimately resulting in seven different types of phosphorylated products (Strahl and Thorner, 2007). The phosphorylation is accomplished by PI kinases (PIKs) and PI phosphate kinases (PIPKs) to generate PI phosphates (PIPs) and bisphosphates (PIP2s), respectively. PI(4, 5)P2 is well known as a second messenger (Doughman et al., 2003; McLaughlin and Murray, 2005). In this study, G-protein-mediated activation of PIKs, PIPKs, and phosphates of PI were also detected (Supplementary Table S8 and Fig. 3).
PIP2 can be either phosphorylated, resulting in PI(3,4,5)P3 or hydrolyzed by a phosphoinositide-specific phospholipase C (PLC) to generate inositol (1,4,5)-trisphosphate (IP3) and 1,2-diacylglycerol (DAG). It is known that PI(3,4,5)P3, through phosphoinositide-specific PLC (EC 3.1.4.11) (Rhee and Bae, 1997), IP3, and DAG function as two important intracellular messengers (Kimmel and Eisen, 1988). Plant and most fungal PLCs belong to the so-called PLCz class, which contain an EF-hand domain and thereby are regulated by Ca2+ through Ca2+ signaling (Munnik and Testerink, 2006, 2009).
However, in this study, PLC could not be detected. This finding is in accordance with the genome sequence analysis of P. sojae and Phytophthora ramorum wherein PIS, PI(P)K, DAG, and phospholipase D (PLD) had been detected but not PLC (Meijer and Govers, 2006). Furthermore, several studies have divulged the importance of PI(3)P for translocation of Phytophthora effectors into host cells, specifically by the binding of RxLR effectors with PI(3)P (Kale et al., 2010; Lu et al., 2013; Yaeno et al., 2011).
Chen et al. (2016) have also reported an enrichment of PI(3)P in P. sojae endoplasmic reticulum and haustorium suggesting its importance in mediating effector translocation and promoting the host infection. Additional evidence has also been provided by Helliwell et al. (2016), who demonstrated the reduction of infectivity of P. tropicalis and P. palmivora, the black pod disease pathogens, by engineering Theobroma cacao to secrete PI(3)P-binding proteins, indicating its potential as a novel target for pathogen control. A recent study by Zhou et al. (2021) linked elevations in Phytophthora PI(3)P levels to virulence; also, targeting of plant-derived antimicrobial peptides (AMPs), fused with “FYVE domain” [a PI(3)P-specific binding domain] to the surface of Phytophthora hyphae resulted in enhanced disease resistance in transgenic soybean and potato lines.
The DAG detected in this study (Fig. 3) can be phosphorylated by diacylglycerol kinase to generate phosphatidic acid (PA), a phospholipid that not only is an intermediate in lipid biosynthesis but also is emerging as one of the most prominent lipid second messengers in eukaryotes including Phytophthora spp. (Testerink and Munnik, 2005). PA can be rapidly and transiently produced after various biotic and abiotic stresses involving the enzyme PLD. Most PLDs contain a C2 (Ca2+ and lipid binding) domain. The C2 domain is thought to be involved in Ca2+-dependent phospholipid binding and thus regulates membrane targeting and PLD activity in a Ca2+-dependent manner (Kolesnikov et al., 2012).
PA can also be produced through hydrolysis of a structural phospholipid, such as phosphatidylcholine, a reaction catalyzed by PLD. PA levels are downregulated by PA phosphohydrolase that dephosphorylates PA, resulting in DAG or the hydrolyzing activity of a PA-specific phospholipase A2, resulting in the signaling molecule lysophosphatidic acid (Meijer et al., 2001).
Ca2+ transporters and a complex set of Ca2+ binding proteins constitute the major molecular components of Ca2+ signaling pathways, exhibiting their role in local and systemic communications. Even very small changes in [Ca2+]cyt are sufficient to activate Ca2+-binding proteins such as calmodulin (CaM) and Ca2+-dependent protein kinases. The concerted action of calcium channels, pumps, and exchangers coordinately regulates the formation of stimulus-specific Ca2+ signatures, which are sensed and transduced by a toolkit of Ca2+ binding proteins that decode the Ca2+ signature into defined phosphorylation or transcriptional changes, thereby leading to downstream physiological responses.
These modulate the activity of a large number of downstream effectors, which finally regulate cellular processes leading, for example, to cell division, expansion, differentiation, or even to cell death, in response to various stimuli such as pathogen attack, symbiosis, or pollen tube growth (Dumas and Gaude, 2006; Harper and Harmon, 2005; Lecourieux et al., 2006). This study revealed the presence of calcium/CaM-dependent protein kinase, CAMK protein kinase, RxLR effector protein, ryanodine-inositol 1,4,5-trisphosphate receptor Ca2+ channel (RIR-CaC) family protein, and serine/threonine-protein phosphatase PP1-beta catalytic subunit (Supplementary Table S8), all of which are well known for their roles in Ca2+ signaling (Berridge et al., 2000; Naveed et al., 2019; Plattner and Verkhratsky, 2013).
Ca2+-permeable channels are usually activated by other factors, such as cAMP/cGMP, [Ca2+]cyt, activation of NADPH oxidases by phosphorylation, and ROS burst (Dodd et al., 2010; Swarbreck et al., 2013). This study also showed the presence of signaling molecules such as cAMP and ROS along with IP3 (Fig. 3), which might have activated Ca2+-permeable cyclic nucleotide-gated channels (CNGCs). Ca2+ signaling in P. palmivora. ROS are produced by respiratory burst oxidase homolog (RBOH) NADPH oxidases, which possess two Ca2+-binding EF-hands. The binding of Ca2+ activates RBOH and, consequently, enhances cellular ROS accumulation (Takeda et al., 2008).
On the contrary, ROS has been shown to activate several Ca2+-permeable channels, such as Shaker-like STELAR K+ OUTWARD RECTIFIER (SKOR) and annexins (Garcia-Mata et al., 2010). The activation of Ca2+-permeable channels then triggers [Ca2+]cyt elevations, which feedback to negatively regulate CNGCs through CaM and at the same time activate Ca2+ pumps such as autoinhibited Ca2+-ATPases (ACAs), thereby terminating influx and enhancing efflux to reduce [Ca2+]cyt levels (Baekgaard et al., 2005).
In oomycetes, cytoplasmic Ca2+ regulates all BP across different life stages (Zheng and Mackrill, 2016). The role of Ca2+ and the activation of IP3 signaling in sporogenesis was observed in P. infestans (Tani et al., 2004) and P. sojae (Hua et al., 2008). PsCAM1, calcium, and CaM-dependent protein-coding genes have also been discovered in P. sojae with a potential role in zoospore formation (Hua et al., 2008). Genome analysis of two oomycetes, P. infestans and Saprolegnia diclina, revealed the involvement of voltage-dependent cation channels activated by cAMP and cGMP in Ca2+ signaling (Verret et al., 2010). In addition, the presence of RIR-CaC family proteins, with roles in Ca2+ signaling in P. infestans and S. diclina, has been demonstrated by Zheng and Mackrill (2016). However, the entire gamut of the Ca2+ signaling pathway in oomycetes is yet to be explored.
Studies have shown the activation of adenylate cyclase (AC) in yeast by Gpa2-mediated Ras2 activation and Ras2-GTP. Gpr1 and Gpa2 are necessary for optimal PKA function (Colombo et al., 2004). However, there is some dispute concerning the level at which Gpa2 acts. Active AC, in turn, stimulates cAMP production, which activates PKA by dissociating the inhibitory cAMP-binding regulatory (R) subunit (Bcy1 in S. cerevisiae) from the catalytic (C) subunits (Tpk1, Tpk2, and Tpk3 in S. cerevisiae).
Active PKA can, in turn, activate other downstream proteins of the MAPK signaling pathway. MAPK signaling includes a three-tiered cascade of protein kinases (Hamel et al., 2012) that elicit many of the responses evoked in cells by changes in certain environmental conditions and upon exposure to a variety of other stimuli such as pathogen, nutrients, and hormones, among others. Many of the components of these pathways and the mechanisms are now known to have been conserved during the evolution of the entire eukaryotic kingdom. Molecular mechanisms by which MAPK cascades operate, propagate signals, and modulate cellular processes are controlled by regulatory factors of both internal and external pathways.
MAPK pathways present in Phytophthora spp. are activated by a common agent, namely, a member of the p21-activated protein kinase (PAK) family of protein kinases, Ste20, similar to yeast (Gustin et al., 1998). Furthermore, the detection of Bmh1 in P. palmivora also might have activated Ste20, one of the major regulator proteins in the MAPK cascade (Dan et al., 2001). Bem1 is an adaptor protein that interacts with proline-rich motifs in Ste20. Ste20 is the Bem1-binding PAK that is activated by Cdc42–GTP and directly phosphorylates Ste11 for the downstream activation of MAPK signaling proteins (Fig. 3), namely Ste7 [mitogen activated protein kinase kinase (MAPKK)] and Fus3 (MAPK). MAPKs are serine/threonine protein kinases that phosphorylate their substrates at –Ser/Thr– Pro– motifs.
Fus3 (Fus3; –TEY–) and Kss1 (Kss1; –TEY–) are the most closely related pair of bona fide MAPKs having their role in the cellular response to peptide pheromones and adjustment to nutrient-limiting conditions, respectively. Fus3 also serves as a negative regulator of filamentous growth; unlike Kss1, it phosphorylates and leads to the degradation of the (Tec1) transcription factor necessary for induction of the genes involved in this developmental stage in the cell cycle, although the molecular basis for this effect seems to be indirect, namely by reducing expression of genes (CLN1, CLN2, and CLB5) encoding cyclins necessary for the G1-S phase transition. MAPK signaling proteins such as Bmh1, Ste20, Ste11, Ste7, Fus3, and Kss1 in P. palmivora might have induced cell division. Similar observations have been reported earlier in pathogenic oomycetes, P. sojae and P. litchii with MAPK proteins such as PsSAK1 and PsMPK7 (Gao et al., 2015a; Li et al., 2010) and PlMAPK2 and PlMAPK2 (Jiang et al., 2018), respectively.
In eukaryotes, cell cycle regulation plays a pivotal role in proper cell division and cellular differentiation (Cooper et al., 2000). The central regulators that govern eukaryotic cell cycle progression are cyclin-dependent kinases and their partners. Proteins and protein regulators of cell cycle such as CDC28, CKS1, MCM2, Cut20/Apc4, condensin family of proteins, double-strand-break repair protein rad21, S-phase kinase-associated protein 1A, and cell division protein kinase were detected in P. palmivora (Supplementary Table S7). Among these, CDC28 was detected in high abundance, a well-known central regulatory protein kinase of the cell cycle in yeast (Lew and Reed, 1993). CDC28 undergoes changes in activity by associating with distinct groups of cyclins that accumulate at different times.
Various cyclin/Cdc28 complexes control different aspects of cell cycle progression, including the commitment step known as START and mitosis. Activation of Cdc28 by G1 cyclins (Cln1, Cln2) helps in the progression of G1 cells to start the mitotic division cycle, thus inducing filamentous growth. Based on the KEGG data analysis and from the available literature, it is proposed that the cell cycle proteins detected in this study could be involved in the normal cell division of fungi and sporulation in P. palmivora (Fig. 3) as described in the case of P. capsici (Ah Fong and Judelson, 2003) and yeast (Neiman, 2011).
P. palmivora proteomic analysis also revealed the detection of elicitins, thioredoxin-like protein, calcineurin-like phosphoesterase putative protein, salicylate hydroxylase putative protein, Crinkler (CRN) family protein, and ABC transporters (Supplementary Table S8). All these proteins exhibit their role in infection to host cells by inducing hypersensitive cell death (Takemoto et al., 2005), reducing the oxidative burst (Resjö et al., 2017), and degradation of salicylic acid (Rabe et al., 2013). Furthermore, the ABC transporters belong to the ubiquitous superfamily of integral membrane proteins and are responsible for the energy-driven translocation of substrates across membranes (Rees et al., 2009). In phytopathogenic fungi, ABC transporters are responsible for transporting toxic phytochemicals produced by the plants upon fungal infection or to provide resistance against the fungicides (Kovalchuk and Driessen, 2010).
The detection of ABC superfamily proteins in P. palmivora (Supplementary Table S8 and Fig. 3) might have a role in overcoming the immune responses of its host plant, as observed in previous studies in P. sojae, P. ramorum, and P. infestans (Kovalchuk and Driessen, 2010). Further studies are necessary to comprehend ABC transporters' detailed mode of action in virulence and drug efflux processes in Phytophthora species. Hence, P. palmivora proteins detected by the LC-MS/MS analysis and annotated using the KEGG database indicate their role in signal transduction reactions through PI, Ca2+, MAPK, and cell cycle pathways suggest the collective involvement of all these pathways and proteins in filamentous growth and sporangia development as reported in related species as well as infection to host plants.
Regulation of autophagy
Autophagy is a catabolic process in eukaryotes to maintain cellular homeostasis under stress and starvation (Chen et al., 2017; Hansen et al., 2018). Autophagy also plays a housekeeping role in removing misfolded or aggregated proteins, clearing damaged organelles, such as mitochondria, endoplasmic reticulum, and peroxisomes, and eliminating intracellular pathogens. Autophagy is a self-eating process in which a remarkable plethora of cargoes can be degraded and recycled by this machinery with high selectivity. The autophagic machinery is sequentially engaged, and the process can be subdivided into distinct steps (Cebollero et al., 2012; Kim et al., 2013; Nair et al., 2010; Nakatogawa et al., 2012; Noda and Inagaki, 2015).
Autophagy-related genes (Atgs) and proteins, along with their role in autophagy-mediated survival, reproduction, and virulence, have also been reported in oomycetes (Chen et al., 2017; Pollack et al., 2009). Genome analysis and reciprocal BLAST analysis in P. sojae led to the detection of 26 Atg genes (Chen et al., 2017). In addition, 13 ATGs, 2 “target of rapamycin” (TOR) genes, and an inhibitor gene (PiFKBP12) coding for FK506 binding protein 12 (an inhibitor of TOR) have been detected in P. infestans (Luo et al., 2014). The presence of active TOR protein negatively regulates autophagy (Cao et al., 2006), and PI3K activates autophagy in P. infestans to play a role in sporulation, zoosporogenesis, and germination of cysts (Lu et al., 2013).
Similar to the autophagy in yeast (Abeliovich and Klionsky, 2001; Rieder and Emr, 1997), this study on P. palmivora also revealed several autophagy proteins, which might exhibit the process by autophagy initiation, nucleation, expansion, fusion with the vacuole, and degradation and recycling of nutrients a given in Figure 4 and Supplementary Table S8. Probably, upon starvation or reduced nutrient availability, autophagy in P. palmivora starts by establishing a phagophore assembly site followed by membrane expansion to form a double-membrane phagophore that surrounds and engulfs cargo destined for autophagy. This leads to the formation of a double-membrane vesicle known as the autophagosome, which is then transported to and fuses with the vacuole for cargo degradation and recycling.
Under nutrient-rich conditions, rapamycin complex 1 (TORC1) is activated to inhibit autophagy, whereas inactivation of this complex in response to stress or starvation leads to autophagy induction (Umekawa and Klionsky, 2012). However, in nutrient-rich conditions, the cargo–SAR–Atg11 complex can activate Atg1 kinase, bypassing the inhibition by TORC1 (Abeliovich and Klionsky, 2001).
Before explaining autophagy-related proteins in P. palmivora, a brief note on the process of autophagy is explained here using S. cerevisiae as a model organism. In brief, actin-related protein Arp2–Arp3 complex, the Atg2 and Atg11 complex, and vesicles containing the integral membrane protein Atg9 initiate nucleation. Formation of the PI(3)K (Class III phosphatidylinositol 3-kinase) complex, including Atg6, VPS15, and VPS34 (vacuolar protein sorting [Vps]), also facilitates initiation of nucleation. PI(3)K complex phosphorylates lipids, gives rise to phosphatidylinositol-3-phosphate, and recruits ATGs to the new membrane.
Then, the phagophore transmembrane protein ATG9 participates in the addition of membrane during elongation and closure of the phagophore to form the autophagosome. Furthermore, a fusion of the autophagosome with vacuole containing hydrolytic enzymes requires the participation of vacuole fusion complex (Rieder and Emr, 1997) through cytoplasm-to-vacuole targeting (Cvt) pathway as observed in yeast (Abeliovich and Klionsky, 2001). Ykt6 is a type of R-SNARE (soluble N-ethylmaleimide sensitive factor attachment protein receptor) protein. In yeast, Ykt6 acts as a key regulator in the autophagosome–vacuole fusion process (Bas et al., 2018). Several autophagy-related proteins were also detected in this study, as shown in Figure 4.
The presence of Ykt6 in P. palmivora probably could play a critical role in the fusion process along with Vps (Vps8 to Vps45) and vacuolar proteases (PEP3, PE4, and PEP5) similar to S. cerevisiae (Kerstens and Van Dijck, 2018; Rieder and Emr, 1997). Hydrolytic enzymes will further degrade the autophagosome and recycle the nutrients. Probably, the recycled nutrients act as the energy source for filamentous growth, sporangia formation, and other normal development of the fungus.
Conclusions
Phytodiagnostics remain an important gap to be addressed in planetary agriculture and systems science research for the early detection of phytopathogens. Progress in this knowledge domain and multi-omics research (Tietel et al., 2021) also have crosscutting valuable impacts including and beyond plant sciences such as new medicinal drug discovery, food, and sustainability in an era when preparing for future ecological crises in the 21st century bear considerable importance.
The LC-MS/MS-based proteomic analysis is one of the important analytical platforms for detecting and quantifying proteins. This study validated many proteins annotated for the genome of P. palmivora. A further search of MS data against the protein databases of closely related species has added many proteins that are yet to be included in the P. palmivora protein database. The proteins identified included the proteins associated with PI, Ca2+, MAPK, cell cycle, and autophagy signal cascade reactions in P. palmivora. The large-scale proteomics data obtained in this study in a hypothesis-free omics systems science research pave the way for new insights on biology, genome annotation, and vegetative growth of the important plant pathogen P. palmivora.
Taken together, the findings and the data presented herein can help for accelerated investigations of host–pathogen interactions, and ultimately, contribute to a foundation for developing future phytopathogen diagnostics and preventive interventions targeting the key signaling molecules of P. palmivora filamentous growth, sporulation, cell cycle, and autophagy.
Footnotes
Authors' Contributions
T.S.K.P. and R.M.K. designed and executed the project. B.N. planned the experiment; data analysis; writing—original draft, figures and tables. M.A. performed the experiment, data analysis, and drew figures. C.N.K. performed a quality check of proteins. R.M.K. wrote the original draft and revised the article. G.K.P. wrote review of literature. M.A.N. performed LC-MS/MS analysis of the sample and acquired data. S.K.M., C.N.K., and S.K. performed data analysis. P.S. V.H., and T.S.K.P. reviewed, edited, and finalized the article. All the authors have read, made intellectual contributions, and approved the submission of the final version of the article.
Acknowledgments
The authors thank Karnataka Biotechnology and Information Technology Services (KBITS), Government of Karnataka, for support to the Center for Systems Biology and Molecular Medicine (CSBMM) at Yenepoya (Deemed to be University), Mangalore, under the Biotechnology Skill Enhancement Programme in Multiomics Technology (BiSEP GO ITD 02 MDA 2017). The authors acknowledge Yenepoya (Deemed to be University) for access to the mass spectrometry instrumentation facility. S.K.B. is a recipient of a Junior Research Fellowship from the Department of Biotechnology-Bioinformatics National Certification (DBT-BINC), Government of India. S.K. and M.A.N. are recipients of Senior Research Fellowships from the Indian Council of Medical Research, Government of India; and the University Grants Commission, Government of India, respectively. M.A. is thankful to Yenepoya (Deemed to be University) for a postdoctoral fellowship.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
The work at ICAR-CPCRI was supported by funding from the Indian Council of Agricultural Research (ICAR-CPCRI Project No. 1000761030).
Supplementary Files
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.