
Abstract
Leptospirosis is one of the most important zoonotic diseases for planetary health. It is caused by Leptospira spp., which poses a formidable challenge in both rural and urban geographies. Discovery of molecular targets is crucial for developing interventions, including vaccines, against leptospirosis. We report here novel systems science insights on Leptospira proteome, posttranslational modifications (PTMs), and pathogen-host interactions, with an eye to bacterial pathophysiology from a functional standpoint. A systematic reanalysis of unassigned spectra from our previous total proteome identification was used for a multi-PTM search. Notably, we identified 3693 unique high-confidence PTM sites corresponding to 1266 proteins (PTM-profiling probability cutoff value ≥75%). The majority of the phosphorylated peptides were found to be GroEL molecular chaperones. Notably, the molecular docking of PTM-GroEL with STAT3, an important signaling protein in cytokine production, resulted in the prediction of druggable “hotspots.” These energetically significant smaller subsets of amino acids (hotspot residues) offer promise for practical applications in planetary health, rational drug design, and peptide engineering. Furthermore, the prediction strategies described here could serve as a starting point for narrowing down the more extensive interface in protein-protein interactions that currently exist. Going forward, systems science approaches and the new insights reported here offer veritable prospects for innovation in preventing and treating leptospirosis.
Introduction
Leptospirosis is one of the most salient zoonotic diseases and a veritable challenge for planetary health. It is caused by Leptospira spp. and is associated with a global burden of the disease of 2.90 million disability-adjusted life years, from 1.03 million cases annually with mortality of 58,900 (Costa et al., 2015; Torgerson et al., 2015). Humans are accidental hosts who get infected from water contaminated with Leptospira from the urine of reservoir hosts such as rodents and other mammals. Currently, there is no effective vaccine available for leptospirosis.
Recent advances in proteomics identified more than 80% of the proteins expressed in Leptospira and revealed its presence in subcellular locations such as the outer membrane, surface, and secretory nature (Sarma et al., 2021; Thoduvayil et al., 2020). However, the mere identification of proteins expressed will not be enough to harness them toward leptospirosis prevention and treatment innovation programs. It is essential to decipher the role of the proteins so that they can be targeted to control Leptospira and its pathogenicity. Different types of posttranslational modifications (PTMs) of proteins play a significant role in orchestrating protein signaling and function as well as serving as a “control gear” in protein function (Wang et al., 2014).
Increasing evidence shows that PTMs play versatile roles in numerous processes such as protein synthesis and its turnover, nitrogen metabolism, cell cycle, dormancy, sporulation, spore germination, persistence and virulence in bacteria (Macek et al., 2019). This shows that proteins bearing PTMs will probably be involved in some functions, and this information, together with their cellular localization and coexpressed molecules, can give better clues toward understanding their biological roles. In addition, protein-protein interactions (PPIs) and host-pathogen interactions can be predicted based on PTMs (Ravikumar et al., 2015). In this regard, investigations on PTMs provide insights into the bacterial pathophysiology and functional consequences during bacterial infection and that can provide a significant contribution in the search for better vaccine candidates.
Major protein PTMs in bacteria are phosphorylation, acetylation, and succinylation. Other types of protein modifications are pupylation and ubiquitin-like modifications, protein glycosylation, and lipidation. Out of many methods used for the detection of PTM, mass spectrometry stands as the frontline tool (Aslebagh et al., 2019; Dunphy et al., 2021). Several mass spectrometry-based studies are conducted at the protein level and, yet, have not focused on PTM biology. The unassigned spectra hitherto acquired from the complete proteome search are used in this study to delve deeper into the PTMs and their role in host-pathogen interaction (Deolankar et al., 2019; Rex et al., 2021). The resolution of protein identification can be enhanced, and their subcellular localization can be discerned by fractionation using Triton X-114, an approach carried out before liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis, as noted elsewhere (Sarma et al., 2021; Thoduvayil et al., 2020).
Materials and Methods
Data sources
The Leptospira label-free total proteome mass spectrometry raw MS/MS data from our previous studies PXD016204 (Thoduvayil et al., 2020) and PXD026044 (Sarma et al., 2021) were reanalyzed for the multi-PTM analysis. These raw files contain both precursor (MS) and fragment masses (MSn), which were searched against the appropriate proteome database to identify peptide spectral matches (PSMs). Not all MS/MS scans are assigned to a peptide sequence of the reference database for various reasons, such as undiscovered sequences, sequence variations, presence of PTMs. The unassigned MS/MS data from the proteome search were retrieved and compared with the reference database using multi-PTMs.
Database search for peptide identification
The MS/MS raw files retrieved from PXD016204 and PXD026044 datasets were searched against the Leptospira interrogans serovar Copenhageni (strain Fiocruz L1-130) protein database downloaded from NCBI RefSeq. The commonly encountered contaminants were added to this database and the searches were performed using SEQUEST-HT and Mascot through Proteome Discoverer (Version 2.2) software suite (Thermo Scientific, Bremen, Germany). Search parameters included trypsin as an enzyme with one missed cleavage. Oxidation (+15.995 Da) of methionine (M) and acetylation (+42.011 Da) of protein N-terminal were set as dynamic modifications. Carbamidomethylation (+57.021 Da) of cysteine (C) was set as a static modification for total proteome analysis.
Unassigned MS/MS spectra obtained from total proteome analysis were subjected to further search against the reference database along with specific PTMs (Deolankar et al., 2019; Pathan et al., 2017). The PTMs such as citrullination (+0.984 Da) of arginine (R); phosphorylation (+79.966 Da) of serine (S), threonine (T), and tyrosine (Y); palmitoylation (+238.230 Da) of cysteine (C); acetylation (+42.011 Da) of lysine (K); and glutarylation (+114.032 Da) of lysine (K) were set as dynamic modifications. Carbamidomethylation (+57.021 Da) of cysteine (C) was set as a static modification.
The mass error for precursor ion and fragment ions was set as 10 ppm and 0.05 Da, respectively. ptmRS node was used to determine the probability of site modification (Taus et al., 2011). The PSMs and peptides that qualified for 1% FDR were considered for further analysis. A parsimonious protein list was generated from the peptide list by grouping proteins in Proteome Discoverer (Version 2.2). The overall workflow of the study is depicted in Figure 1.

The workflow of PTM analysis: label-free MS/MS raw files were downloaded from the PRIDE archive and searched against Leptospira interrogans serovar Copenhageni (strain Fiocruz L1-130) protein database in Proteome Discoverer software suite 2.2 with selected PTMs using SEQUEST-HT and Mascot. PTM-Pro tool was used to find the high-confidence PTMs, and these modified proteins were subjected to various bioinformatics-based analysis. PTM-Pro, posttranslational modification-profiling.
Bioinformatics analysis
Posttranslational modification-profiling (PTM-Pro) (Version 2.0) (Patil et al., 2018), an online tool, was used to fetch high-confidence PTMs, which requires peptide sequence, protein group accession, and ptmRS probabilities as input files. A probability cutoff of 75% with Leptospira interrogans as the reference organism was applied as parameters. Gene ontology (GO) analysis of the Leptospira interrogans was carried out using the PANTHER tool (Mi and Thomas, 2009). Subcellular localization of leptospiral proteins predicted using CELLO v.2.5 (http://cello.life.nctu.edu.tw/) for five localization sites—cytoplasmic, inner membrane, periplasmic space, outer membrane, and extracellular space—were also included for all the fractions (Yu et al., 2006).
Host-pathogen PPI
To find the identification and functional role of host-pathogen interaction, we used sequence-based PPI prediction tool HPIDB 3.0 (Host-Pathogen Interaction Database) (https://hpidb.igbb.msstate.edu/sequence.html). Modified proteins identified from multi-PTM search (FASTA sequence) were used as the input file. The search parameters for HPIDB 3.0 were BLOSUM 62 as matrix, 0.00001 as e-value, 60% identity filter, and 60% query coverage filter and searched for the top match (Ammari et al., 2016).
Protein structure retrieval, PTM modeling, and refinement
The X-ray crystallographic structure of Human STAT3 was retrieved from Protein Data Bank (PDB ID: 6NUQ). The FASTA sequence of Leptospira interrogans chaperonin GroEL was retrieved from the UniProt database (ID: P61438). The GroEL sequence was modeled into a 3D structure using highly accurate protein structure predictor “AlphaFold 2” using AI for scientific discovery (Jumper et al., 2021). Both STAT3 and GroEL structures were preprocessed in the Schrodinger suite's “Protein preparation Wizard” for missing hydrogen atoms, bond order deviations, sidechain fixation, and loop refinement before energy minimization using an OPLS3e force field (Schrödinger, LLC, New York, NY, 2019-1). The processed proteins were further validated using the Ramachandran plot's Phi/Psi distribution.
PTMs from GroEL's pull-down protein spectra were substituted at the desired positions of the amino acid residues for PTMs. The amino acid residues Ser 227 and Thr (175, 269, 295,311, and 323) were phosphorylated by adding a phosphate group in monoanionic protonation states. For acetylation, an N-acetyl group was substituted at Lys 57 and 471. For all substitutions, the “Vienna-PTM 2.0” webserver was utilized. The modeled posttranslationally modified GroEL (PTM-GroEL) was refined and validated using a process identical to that was used for STAT3 and GroEL.
Because PTMs might cause conformational changes in native protein side chains, the modeled PTM-GroEL was subjected to a 100 NS molecular dynamic simulation (MD Simulation) using the TIP3 water system. Before running the MD simulation, the charges were neutralized with extra counter ions (Na+) in 0.15 M NaCl. At 300K temperature and 1.01325 bar pressure, the simulation was run with the OPLS3e force field and an NPT ensemble. Following the MD simulation, the 1000th frame of the MD trajectory was extracted and processed for protein refinement using the same methods as previously reported. To complete all of the MD simulation phases, the academic version of DESMOND 2.4 was used. Further protein-protein docking investigations were conducted using the refined structures of GroEL, STAT3, and PTM-GroEL.
Protein docking
Before docking, the amino acids that comprise the Human STAT3 domain region were gathered from published literature and curated sources. Based on this, the STAT3 amino acid residues 580 to 670 were added as restrictions for GroEL and PTM-GroEL binding. GroEL and PTM-GroEL were not subjected to any binding limitations, allowing both pathogenic proteins to bind to the binding site of Human STAT3 in any possible pose. The protein-protein docking was carried out using the ZDOCK 3.0.2 website (https://zdock.umassmed.edu/). Based on Z-score, the top 10 anticipated complexes for STAT3/GroEL and STAT3/PTM-GroEL were investigated separately on their interacting partner utilizing an interactive monitor incorporated in the BIOVIA Discovery Studio (DS) visualizer.
Identification of druggable binding sites of STAT3/PTM-GroEL protein complex
The druggable target binding sites and “Hot spot” residues in the PTM-GroEL/STAT3 PPI complex was evaluated based on the changes in solvent accessible surface area (ΔSASA) of each side chains along with its binding energy using ANCHOR webserver (http://structure.pitt.edu/anchor/). This webserver facilitates the identification of important “Hot Spot” residues for small molecule inhibition.
Results
For the multi-PTM analysis, we utilized the total proteome results from our previous studies, (1) Triton X-114 fractionated subcellular proteome (aqueous, detergent, and pellet) (PXD016204) and (2) extracellular proteome analysis (supernatant and wash) (PXD026044). The unassigned spectra obtained after comprehensive proteome search (aqueous, detergent, pellet, supernatant, and wash) was filtered and searched for multi-PTMs such as citrullination (R), phosphorylation (S, T and Y), palmitoylation (C), acetylation (K), and glutarylation (K). This led to the identification of 1606 protein groups in multi-PTM and 1266 proteins in PTM-Pro (Supplementary Table S1). The multi-PTM presence, identified by PTM-Pro, in leptospiral proteins is depicted in Figure 2, and the list of proteins are provided in Supplementary Table S2.
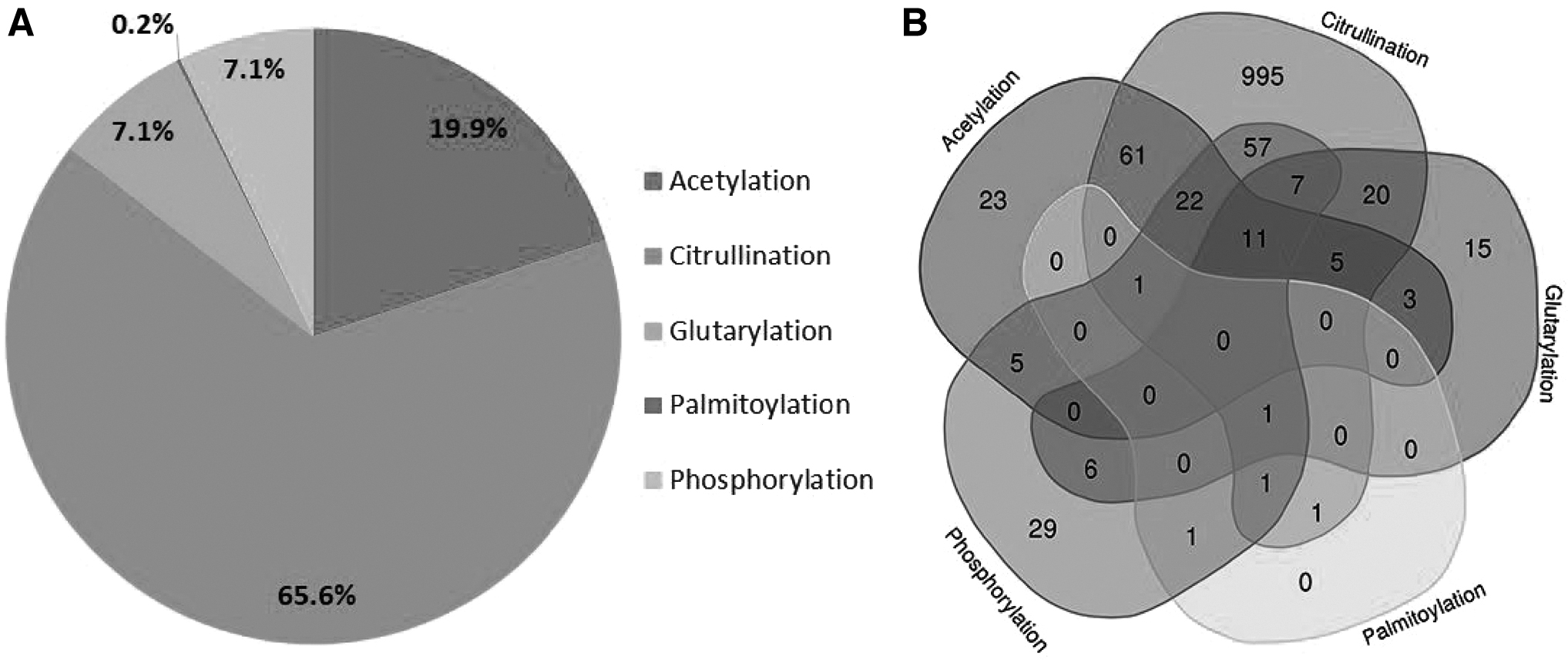
PTM profile of Leptospira:
Posttranslational modification-profiling
A total of 3693 unique high-confidence PTM sites corresponding to 1266 proteins (PTM-Pro probability cutoff value is ≥75%) have been identified. These PTMs include 2911 unique citrullination sites corresponding to 1183 proteins. The PTM-Pro showed that the major modification was citrullination, accounting for 65.6% with 995 uniform modifications and the remaining 187 as part of multiple modifications. Similarly, acetylation forms the second major modification (19.9%), followed by glutarylation (7.1%), phosphorylation (7.1%), and palmitoylation (0.2%) as shown in Figure 2.
Phosphorylation
In Leptospira, phosphorylation plays a vital role in the regulatory mechanism of cellular activity, controlling functions such as growth, intracellular membrane traffic, cell cycle, apoptosis, biofilm formation, and response to stress or external stimuli.
We identified 203 unique phosphorylation sites corresponding to 141 proteins. These phosphorylation sites include serine (S) (92 sites), followed by threonine (T) (80 sites) and tyrosine (Y) (31 sites). We identified that 18 proteins were hypothetical, and 6 proteins were antisigma factor antagonists, as well as proteins such as molecular chaperones (GroEL, GroES, HtpG, DnaJ), lipoproteins (LipL36, LipL41, LipL45, LipL46, LipL71), and domain of unknown function proteins (DUF1987, DUF1554, DUF3095, DUF1420), which were phosphorylated. Most of the phosphorylated proteins identified with PTM play an essential role in the metabolic processes.
Acetylation and glutarylation: lysine modification
Lysine PTMs are the most important player in regulating diverse functions of proteins and biological processes such as DNA replication and gene regulations. In the current study, we have identified different types of PTMs on lysine, namely acetylation and glutarylation.
Acetylation
In bacteria, the acetylation of proteins is involved in PPIs, DNA-protein interactions, metabolic processes, and adaptation to environmental changes (Macek et al., 2019). This study identified 151 unique acetylation sites corresponding to 131 proteins. We found that there were 11 hypothetical membrane proteins, including PIN/TRAM domain containing protein, fibronectin type III domain-containing protein (WP_000836523.1), cytoplasmic protein flagellar assembly protein FliH (WP_001096786.1), as well as molecular chaperone (GroEL, DnaK) protein ClpB (WP_000762669.1), metabolism-related proteins such as malate dehydrogenase (WP_000048017.1), phosphoenolpyruvate carboxykinase (ATP) (WP_001148872.1), phosphoglycerate kinase (WP_000422649.1), pyruvate kinase (WP_000698533.1), and acyl-CoA dehydrogenase (WP_001199925.1), which were acetylated.
Glutarylation
The proteins that are glutarylated has an essential role in cellular functions like metabolism, translation, cellular localization, and protein folding (Xie et al., 2016). We identified 80 unique glutarylation sites corresponding to 73 proteins. There were 16 hypothetical proteins, as well as molecular chaperones (HtpG, DnaK), NADH-quinone oxidoreductase subunit D (WP_000990075.1), helicase (WP_000063951.1), and DNA gyrase subunit B (WP_000102225.1), which were glutarylated.
Palmitoylation
Palmitoylation impacts PPIs with membranes, membrane compartmentalization, trafficking, and stability (Sobocinska et al., 2017). Here, we identified five unique palmitoylation sites corresponding to five proteins. The proteins such as ketol-acid reductor isomerase (WP_001284348.1), electron transfer flavoprotein subunit alpha/FixB family protein (WP_000072965.1), UDP-glucose 4-epimerase (WP_001067557.1), DUF1554 domain-containing protein (WP_002084608.1), and adenylate/guanylate cyclase domain-containing protein (WP_001291001.1) were found to be palmitoylated. Table 1 enlists the modified proteins that were commonly found in our study. It includes the identified posttranslationally modified proteins, peptides, and site modifications.
List of Posttranslationally Modified Proteins, Peptides, and Site Modifications Identified from the Study
GO and pathway analysis
To better understand the function of modified proteins, they were classified according to related GO terms and pathway analysis by PANTHER (Fig. 3). To interpret GO characteristics effectively, each GO class analyzed the PTM proteins with their number (number of unique proteins) and quantity (assessed through the iBAQ values of proteins). It was found that within major GO classes such as molecular function and biological process showed a higher abundance of PTM proteins than in cellular component protein class and pathway. On a detailed analysis of each major class, the subclasses structural molecule activity, biological regulation, structural protein, and pyruvate metabolism showed a high abundance of PTM proteins (Fig. 3A).
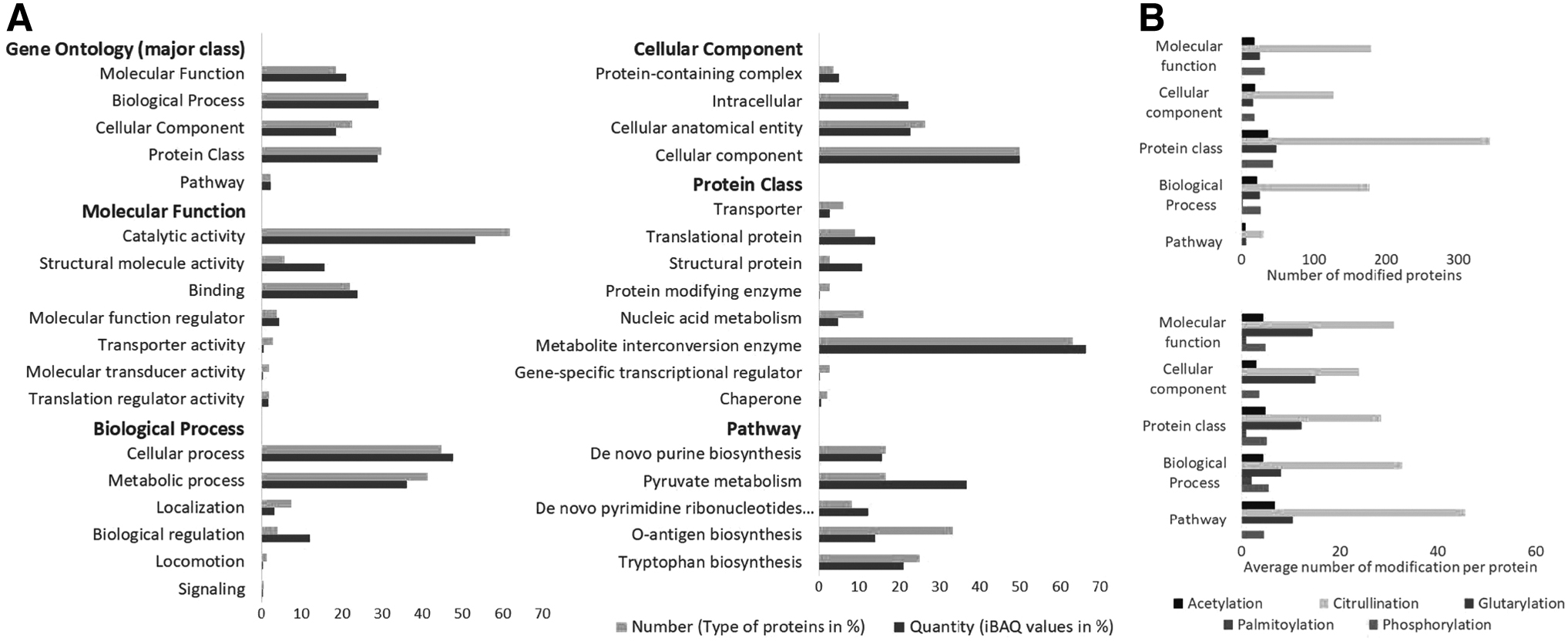
GO analysis of modified proteins: The modified proteins from all the fractions were subjected to GO analysis using PANTHER. The bar chart represents GO annotations in five major classes: Molecular function, Biological Processes, Cellular component, Pathway, and Protein class
Further analysis of subgroups in GO and their PTMs that illustrate both ways PTM proteins and their GO properties and PTM proteins associated with each GO group are shown in Supplementary Figure S1A. The analysis on the type of modification in each GO group showed that the highest modification is citrullination in protein class; however, highest modification per protein was found as citrullination in proteins of the pathway GO group. Then, the GO groups of molecular function and cellular component, the glutarylation forms the highest modification per protein, as shown in Figure 3B.
Subcellular localization prediction
The prediction on localization of modified leptospiral proteins was carried out using CELLO v.2.5 (Supplementary Fig. S1B). Most of the proteins found were cytoplasmic and outer membrane proteins in all the fractions. The results of the prediction are given in Supplementary Table S3.
Host-pathogen PPI network mapping
To get a deeper insight into the cellular processes influenced by the pathogen interaction with the host, we mapped a PPI network represented in Supplementary Figure S2. Our result shows that one of the leptospiral proteins, UvrABC system protein A (WP-001156251.1), interacts with CD4 (coreceptor for the T cell receptor), NFKB1 (controls transcription of DNA, cytokine production), GSTP1 (role in detoxification), and COL4A1 of the host. Furthermore, the result obtained from HPIDB 3.0 showed PPIs between 19 leptospiral proteins and the human host. The 19 leptospiral proteins contain significant PTMs and shared GO (GO_Slim) between 14 proteins, shared COG (Depository of Live Systems Domain Analyzer) between 4 proteins, shared features (UniProt) between 10 proteins, and shared function (UniProt) between 13 proteins of the orthologs proteins from the pathogen that used to identify similarity in interaction using HPIDB 3.0 (Supplementary Table S4).
Molecular docking and interaction analysis
GroEL/STAT3 and PTM-GroEL/STAT3
The top 10 predicted complexes for GroEL/STAT3 and PTM-GroEL/STAT3 from ZDOCK data were examined in depth in DS visualizer based on binding mode and interaction residues. As a result, Complex-2 of GroEL/STAT3 and Complex-3 of PTM-GroEL/STAT3 were found to have favorable hydrogen, electrostatic, and hydrophobic bond interactions. As a result, each of the complexes above was picked as an appropriate model for further investigation.
PTM-GroEL residues Asp216, Phe218, Gln241, Gly243, Lys244, Pro245, Lys268, TIP269, Thr312, Leu313, and Arg318, interacted with STAT3-binding pocket residues Tyr640, Gln644, Met648, Glu652, Met655, Lys658, Met660, and Leu666 in Complex-3 (Fig. 4). TIP269 (phosphorylated tyrosine residue) forms two hydrogen bond interactions with Lys658 of the Human STAT3 protein, which is intriguing. This shows that the interplay between Lys658 and STAT3 signaling is essential. In addition, the PTM-GroEL interaction pattern reveals potential STAT3 activation binding sites in PTM-GroEL. In Complex 2, hydrogen, hydrophobic, and electrostatic interactions were observed between GroEL amino acid residues Pro201, Tyr202, Ser228, Glu256, and Ala259, and STAT3-binding pocket residues Val637, Glu638, Tyr640, Tyr657, Lys658, and Leu666. The nature and types of interactions between the two complexes are summarized in Tables 2 and 3.

Binding mode of PTM-GroEL/STAT3 structural complex and its close-up view of intermolecular interactions.
Interacting Amino Acid Residues of PTM-GroEL/STAT3 and GroEL/STAT3 Complexes Predicted by Z-Dock Webserver
Major contributing residues are shown in boldface.
PTM, posttranslational modification.
Protein-Protein Interactions in the Proposed PTM-GroEL with Human STAT3
The ANCHOR webserver was used to identify druggable binding sites for the PTM-GroEL/STAT3 PPI complex. The most significant residues at the PTM-GroEL/STAT3 interaction interface were identified as Met648, Leu666, Met660, Gln643, Tyr657, Leu313, Lys268, and Ala242 (Fig. 5). The STAT3 residues Met648, Leu666, Met660, and Tyr657 showed the highest ΔSASA and the lowest estimated binding energy (i.e., favorable) (Table 4).
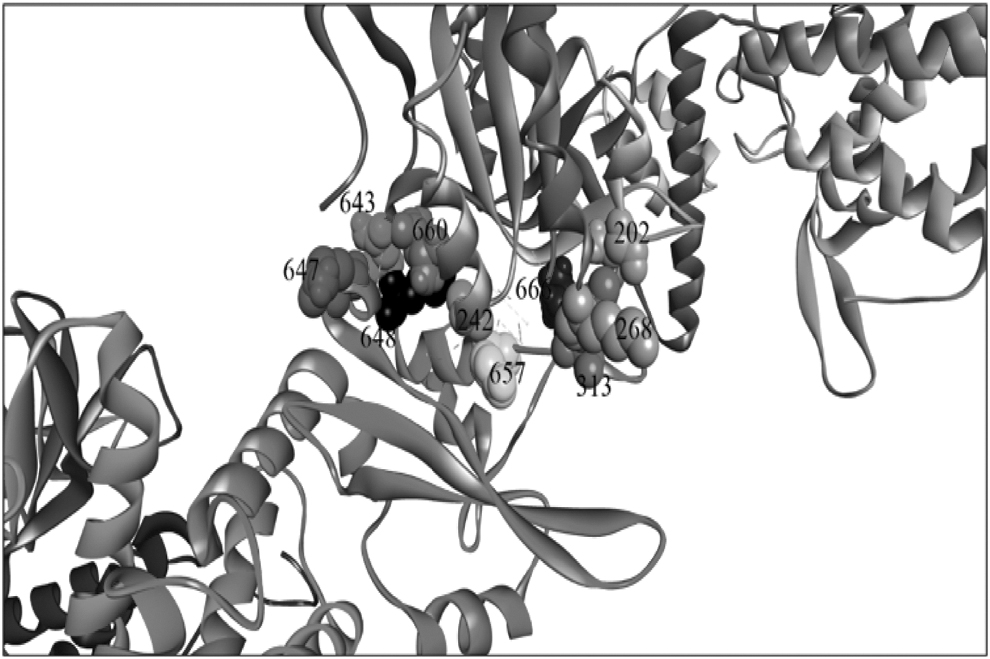
Potential binding sites in the PTM-GroEL/STAT3 complex presented as occupancy in CPK style in the structure.
Potential Binding Sites in the PTM-GroEL Complex Along with its Solvent Accessible Surface Area and its Binding Energy Using ANCHOR Webserver
ΔSASA, Solvent Accessible Surface Area.
TIP269 hydrogen bond interactions with Lys658 also demonstrate the relevance of Lys658 in STAT3 activation. Panaxadiol, a triterpenoid, has been shown to prevent pancreatic cancer growth via STAT3 signaling, with its inhibitory activity mediated, in part, by interactions with Met468 and Lys658 (Fan et al., 2021). SD-36, a novel selective small-molecule STAT3 degrader, was discovered to work via a water-mediated hydrogen bond interaction at Lys658 and hydrophobic occupancy of the diphenyl methyl group at Leu666 and Met660 (Bai et al., 2019).
Based on the findings, it is feasible to conclude that the mapped (Met648, Leu666, Met660, and Tyr657) sites were druggable “Hotspots” that might be used to develop small-molecule inhibitors that disrupt PPIs between PTM-GroEL and STAT3. Supplementary Figure S3A illustrates the GroEL protein sequence and peptides and the typical spectra found in the study.
Data availability
The mass spectrometry proteomics data have been deposited to the Proteome Xchange Consortium (www.proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD030370.
Discussion
Proteomic analysis used in this study was high-resolution LC-MS/MS that made highest protein detection ever performed in Leptospira that identified 82% of unmodified protein from the transcriptome of Leptospira interrogans serovar Copenhageni strain Fiocruz L1-130 reported in NCBI database (Sarma et al., 2021; Thoduvayil et al., 2020). In this study, a separate analysis specifically targeted identifying protein modifications using PTM-Pro tool and found 1266 proteins are modified, which account for 35% of the transcriptome of the Leptospira strain. PTMs play a significant role in PPIs (Duan and Walther, 2015).
It was found that citrullination forms the most significant number of PTMs in Leptospira. Citrullination plays an important role in the structural stability of proteins, PPIs, formation of hydrogen bonds, and can cause protein denaturation (Knuckley et al., 2010; Tarcsa et al., 1996). In addition to these, citrullinated antigens are found to aggravate the pathological signs of arthritis (Jung et al., 2017) and modify the contact point of T cell receptors that alter the balance between STAT5 and STAT3 activation in responding T cells (Tibbitt et al., 2016). Citrullination was found in leptospiral proteins identified in all GO classes, which indicates their involvement in various biological functions.
The second highest PTM was acetylation, which plays an essential role in bacterial virulence by influencing the activities of bacterial metabolism, chemotaxis, DNA replication, and many other cellular processes (Ouidir et al., 2016; Ren et al., 2017), which is evidenced in the presence of acetylated proteins in many GO functions cellular component and protein classes of similar functions. Phosphorylation can influence folding, stability, and activity and control PPIs (Mijakovic et al., 2016). Our results showed high expression of several phosphorylated proteins, including lipoprotein LipL36 (WP_001253849.1), a virulence factor expressed under pathogenic conditions (Fraser and Brown, 2017).
Similarly, an antisigma factor antagonist (WP_000406878.1) that contains sulfate transporter and antisigma factor antagonist (STAS) domain, which can control antisigma factors and liberate sigma factors to support transcription of target genes or operons that help the pathogen for its survival (Sharma et al., 2011). This shows the significance of phosphorylated proteins in Leptospira. Highly abundant glutarylated proteins molecular chaperone HtpG (WP_001288005.1) is a virulence factor (King et al., 2014) and MULTISPECIES: OmpA family protein (WP_000239305.1), an ortholog of this protein, Loa22, in L. interrogans serovar Lai, is found to be essential for virulence (Ristow et al., 2007). This shows that these highly abundant glutarylated proteins are associated with virulence and can target antileptospirosis strategy.
The PPIs found between 19 leptospiral proteins with PTM and human host as identified by HPIDB 3.0 showed that many features, GO, COG, and functional properties between their orthologous pathogen add probability and importance to the interactions. The illustration of the interaction between GroEL/PTM-GroEL and STAT3 is only an example here. Even though GroEL is mainly known as a chaperonin that expresses under heat shock conditions, it is imperative due to its critical role in protein folding and many other activities collectively described as “moonlighting” functions (Kupper et al., 2014).
Leptospira infects from contaminated water sources to humans thriving a temperature range of environmental water temperature around 25°C to 37°C and osmolarity from 67 mOsM of freshwater to 300 mOsM of body fluids, which is enormous stress that shows the importance of GroEL in Leptospira. The GroEL is the significant molecule that manages such stress and establishes successful infection and pathogenesis (Fourie and Wilson, 2020). The host-pathogen protein interaction listed here are much more critical, and further study may lead to the development of drug and vaccine candidates.
The comparison of the identified phosphopeptides and acetylated peptides with the enriched study by Cao et al. (2010), resulted in the identification of four common phosphopeptides and one common acetylated peptide with the same site modifications (Cao et al., 2010) (Supplementary Fig. S3B, C). DUF1987 domain-containing protein (T8 site), antisigma factor antagonist [WP_000884017.1] (S12 and S13 site), antisigma factor antagonist [WP_000406878.1] (S7 site), and antisigma factor antagonist [WP_000448574.1] (S4 site) are the common phosphopeptides identified. Common acetylated peptide identified is of PIN/TRAM domain-containing protein [WP_000581182.1] (K3 site).
Conclusions
We report a list of modified proteins involved in Leptospira pathogenesis that offers new insights. PTMs that make changes in the functional characteristics and its capacity to interact with host proteins have a remarkable impact on pathogenicity and survival of the pathogen in the host. However, in-depth knowledge in this regard is yet to be deciphered. The present report shows the influence of PTMs in pathogen-host protein interactions. Further functional analysis of the resulting protein complexes can reveal the significance of micromanipulation of PTMs on the interacting milieu of the pathogen and host protein.
The adaptiveness of the change and resulting interaction can offer a precise idea of how they can be incorporated in vaccine and drug design in the future. Also, the prediction strategies described here could serve as a starting point for narrowing down the more extensive interface in PPIs that currently exist. Going forward, systems science approaches and the new insights reported here offer veritable prospects for innovation in preventing and treating leptospirosis.
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
The authors are grateful to the Indian Council of Medical Research, New Delhi, India (grant no. Leptos/22/2013-ECD-1) to M.G. Madanan, and the Indian Council of Medical Research, New Delhi, India, Senior Research Fellowship (grant no. ICMR-SRF 2020-0756/PROTEOMICS-BMS) to H. Phukan.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.