
Abstract
Cardiovascular disease (CVD) is the leading cause of death among adults in developed countries. Among CVDs, abdominal aortic aneurysm (AAA) and aortic occlusive disease (AOD) are of great public health importance because of the high mortality rate in the elderly population. Despite significant molecular insights into AAA and AOD, the molecular mechanisms of these diseases remain unclear, and the current lack of robust diagnostic and prognostic biomarkers requires novel approaches to biomarker discovery and molecular targeting. In this study, we performed a comparative analysis of genome-wide expression data from patients with large AAA (n = 29), small AAA (n = 20), AOD (n = 9), and controls (n = 10). Specifically, we identified the differentially expressed genes and associated molecular pathways and biological processes (BPs) in each disease. Using a systems science approach, these data were linked to comprehensive human biological networks (i.e., protein-protein interaction, transcriptional regulatory, and metabolic networks) to identify molecular signatures of the salient mechanisms of AAA and AOD. Significant alterations in lipid metabolism and valine, leucine, and isoleucine metabolism, as well as neurodegenerative diseases and sex differences in the pathogenesis of AAA and AOD were identified. In the presence of aneurysm, size-dependent changes in lipid metabolism were observed. In addition, molecules and signaling pathways related to immunity, inflammation, infectious disease, and oxidative phosphorylation were identified in common. The results of the comparative and integrative analyzes revealed important clues to disease mechanisms and reporter molecules at various levels that warrant future development as potential prognostic biomarkers and putative therapeutic targets.
Introduction
Cardiovascular disease (CVD) is the leading cause of death in adults in developed countries (Peshkova et al., 2016). Among CVDs, abdominal aortic aneurysm (AAA) refers to an irreversible dilatation of the abdominal aorta between the diaphragm and the iliac bifurcation, and usually involves all three layers of the vascular wall. AAA is characterized by an enlargement of arterial diameter by at least 1.5 times (Toghill et al., 2017). AAA is a common condition with public health significance. One meta-analysis of 53 studies of AAA found an overall pooled prevalence of 4.8% in the general population (Li et al., 2013).
Proliferation of intimal smooth muscle cells in the surrounding connective tissue and accumulation of lipids are among the hallmarks of aortic occlusive disease (AOD), which refers to the obstruction of the aorta. AOD could be triggered by atherosclerosis, in which plaque formation is associated with apoptosis of vascular smooth muscle cells, endothelial cells, and inflammatory cells. In addition, genetic factors also contribute to the development of AOD (Armstrong et al., 2002; Lucas et al., 2016).
Atherosclerosis is another notable CVD risk factor (Toghill et al., 2017). While patients with AAA develop arterial dilation, AOD results in arterial occlusion. Thus, both diseases lead to atherosclerotic burden in the aorta (IJpma et al., 2019). Atherosclerosis is thought to be the main reason of aneurysmal changes in the aorta. The reduced oxygen flow to the aortic wall due to atherosclerotic plaques leads to wall deterioration, and the accumulation of these plaques can lead to aneurysmal dilation in some patients and occlusive disease in others (Koksal et al., 2007; Shteinberg et al., 2000).
Several genetic, hemodynamic, and environmental factors together with lifestyle, age, gender, smoking, and diet contribute to the progression of AAA and AOD (Johnsen et al., 2010; Peshkova et al., 2016; Shteinberg et al., 2000). Inflammation is among the main pathological hallmarks of both diseases (Armstrong et al., 2002). Although AAA and AOD share similar risk factors and mechanisms of pathogenesis, recent studies suggest that their pathologies also display divergence. For example, AAA is not always a manifestation of AOD (IJpma et al., 2019; Toghill et al., 2017). Therefore, the elucidation of the diversity of molecular mechanisms responsible for AAA and AOD would increase our understanding of both diseases' pathogenesis and provide valuable information for predictive biomarker discovery and development.
Clinical context on AAA and AOD are also noteworthy in regard to future diagnostics research. AAA is a multifactorial disease whose progression is mostly asymptomatic and no effective drug therapy has been developed so far. The progressive enlargement of abdominal aorta may result in an aortic rupture, which often leads to death (Golledge, 2019; Moris et al., 2014). Although in-hospital mortality of ruptured AAA is about 40%, the total mortality, including pre-hospitalization deaths, is approximately 60–80% (Kühnl et al., 2017). It is estimated that the ruptured aneurysm accounts for 150,000–200,000 deaths every year worldwide, and the risk of aortic rupture is greater for large aneurysms when compared to small ones (Golledge, 2019). Reducing the mortality rate due to rupture can be achieved, however, with early diagnosis and precision/personalized treatments (Ciborowski et al., 2012).
Currently, early diagnosis of AAA is generally attained by incidental imaging or screening programs (Golledge, 2019). Imaging techniques, including computed tomography angiogram, ultrasonography, and magnetic resonance imaging, are used for AAA screening (Moris et al., 2014). Ultrasonography-based screening programs that were carried out in various centers revealed a substantial reduction in AAA-related deaths. Despite the efficacy of screening programs, diagnosis largely depends on the physician experience, and the selected method. Alternative diagnostic methodologies, particularly noninvasive biomarkers in the peripheral blood, would be invaluable to overcome the difficulties of screening-based methods (Moris et al., 2014; Zhang et al., 2015).
The current lack of robust diagnostic and prognostic biomarkers, not to mention the morbidity and mortality risks associated with AOD and AAA rupture, calls for new approaches for discovery of biomarkers and molecular targets.
In recent years, high-throughput technologies have been extensively deployed to discover systems diagnostics for common and rare human diseases, leading to diagnostic successes and new therapeutic strategies. For example, previous studies based on gene expression profiling revealed thousands of differentially expressed genes (DEGs) in AAA and AOD (Biros et al., 2015; IJpma et al., 2019; Meng et al., 2021). However, these existing data are waiting for multiomics data integration to harness their full potential as systems diagnostics.
The reporter features approach is an efficient data integration strategy that integrates gene expression profiles with biomolecular networks (i.e., metabolic, transcriptional regulatory, and protein-protein interaction (PPI) networks) to identify biological features such as biological processes (BPs), signaling and metabolic pathways, and biomolecules such as receptors, transcription factors (TFs), miRNAs, and metabolites around which significant changes related to pathogenesis occur (Oliveira et al., 2008; Patil and Nielsen, 2005).
The applicability of this strategy in identifying diagnostic and prognostic biomarkers and potential drug targets has been demonstrated in several diseases such as ovarian cancer (Gov et al., 2017), head and neck cancer (Islam et al., 2018), cervical cancer Kori and Arga, 2018), renal cell carcinoma (Caliskan et al., 2020), papillary thyroid cancer (Gulfidan et al., 2022), and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (Moolamalla et al., 2021).
We report here an original investigation and comparative analysis of genome-wide expression data from patients with large AAA, small AAA, and AOD and controls. Specifically, we compared DEGs, and identified the associated molecular pathways, and BPs. Using a systems science approach, these data were linked to comprehensive human biological networks to identify molecular signatures of the salient mechanisms of disease in AAA and AOD. PPI networks were reconstructed for all disease states and topological analyses of these networks revealed hub proteins. Integrative analysis of gene expression data with a genome-scale metabolic model, a PPI network, and a transcriptional regulatory network identified reporter biomolecules (metabolites, receptors, and TFs, respectively) that warrant future development as potential prognostic biomarkers and putative therapeutic targets.
Materials and Methods
This article presents a computational analysis of gene expression data obtained from public databases. It does not contain any studies with human and animal subjects and therefore it does not require IRB approval.
Gene expression dataset
To gather AAA- and AOD-associated gene expression profiling datasets, we comprehensively screened the publicly available NCBI Gene Expression Omnibus (GEO) database (Barrett and Edgar, 2006). Among transcriptome datasets, gene expression data with accession number GSE57691 (Biros et al., 2015) were employed considering the presence of aortic specimens without any treatment and its large sample size (58 disease and 10 control aortic specimens), and regarding AAA patient groups as small and large AAA.
The samples included 58 aortic specimens from 20 patients with small AAA, 29 patients with large AAA, 9 patients with AOD, and 10 control aortic specimens obtained from organ donors. The mean values of aortic diameters were 54.3 ± 2.3 mm, 68.4 ± 14.3 mm, and 19.6 ± 2.6 mm for small AAA, large AAA, and AOD, respectively. A total of 40% of the subjects in the control group was female, and the proportion of females was lower in patients enrolled (0% in small AAA, 7% in large AAA, and 11% in AOD). The mean ages were 68.8 ± 6.9, 70.5 ± 7.1, 61.6 ± 9.3, and 68.4 ± 4.5 years for small AAA, large AAA, AOD, and control subjects, respectively.
Identification of DEGs
The raw data were normalized using quantile normalization and suitability and cross-comparability of samples for differential expression analysis was checked using median-centered expression values generated by GEO2R tool (www.ncbi.nlm.nih.gov/geo/geo2r/). Comparisons were performed for the following cases: (1) large AAA versus control, (2) small AAA versus control, and (3) AOD versus control. DEGs were identified using Linear Models for Microarray Data (LIMMA) method (Smyth, 2004). The Benjamini–Hochberg method was used to control the false discovery rate (FDR). An adjusted p-value threshold of 0.05 (adjusted-p < 0.05) was applied to determine the statistical significance, and a cutoff of 1.5 was used for fold change to determine upregulated and downregulated genes.
Reconstruction and analysis of PPI networks
Physical PPIs among DEGs were extracted from the BioGRID database (release 3.4.161) (Oughtred et al., 2019), which contains 35,688 physical experimentally detected PPIs among 8570 human proteins. PPI networks were individually reconstructed around the upregulated and downregulated DEGs considering three cases. The topological properties of the networks were determined using the Cytohubba (Chin et al., 2014) plug-in of Cytoscape (v3.5.0) (Shannon et al., 2003). The dual metric approach (Karagoz et al., 2015) considering degree and betweenness centrality metrics simultaneously was used to identify hub proteins. The top 10 proteins in the PPI network with the highest degree and betweenness centrality values were determined to be hub proteins.
Diagnostic performance analysis of the hub proteins
The gene expression profiles of the hub genes were extracted from the GSE57691 dataset (Biros et al., 2015), and principal component analysis (PCA) was performed based on the gene expression profiles of hubs (22 hubs for large AAA and for 20 hubs for small AAA and AOD). The first three principal components, which explain the highest variances, were considered in determining the sensitivity (the proportion of positive test results in patients) and specificity (the proportion of negative test results in healthy individuals) (Kori et al., 2019) to validate the diagnostic performance of the hub proteins.
Identification of reporter metabolites
The metabolites around which the most significant transcriptional changes occurred were determined by reporter metabolite analysis. The Human Metabolic Reaction model (HMR 2.0) (Mardinoglu et al., 2014) is from Human Metabolic Atlas (Pornputtapong et al., 2015) and was integrated with gene expression data from large AAA, small AAA, and AOD using the Reporter Metabolites tool implemented in BioMet Toolbox (v2.0) (Garcia-Albornoz et al., 2014). The adjusted p-value threshold of 0.05 (adjusted-p < 0.05) was used for the determination of reporter metabolites. Enrichment analyses of reporter metabolites were performed using the server Metabolites Biological Role (MBRole) (v2.0) (López-Ibáñez et al., 2016), and pathways with FDR corrected p-value <0.05 were considered statistically significant.
Identification of reporter TFs and receptors
The reporter TFs and receptors were identified by integrating gene expression data from large AAA, small AAA, and AOD with the transcriptional regulatory network with TF-target gene interactions and receptor-protein interactions. The experimentally validated TF-target gene interactions were obtained from the human transcriptional regulatory interaction network (Gov and Arga, 2016) and the Human Transcriptional Regulation Interactions database (HTRIdb) (Bovolenta et al., 2012). The proteins with receptor activity (GO: 0004872) were extracted from DAVID (Huang et al., 2009), PANTHER (Mi et al., 2021), and GeneCodis (Tabas-Madrid et al., 2012) databases, and the physical interactions of these receptors were extracted from the human PPI network.
The reporter features algorithm (Patil and Nielsen, 2005) was adapted as previously described (Kori and Arga, 2018) and implemented in MATLAB (R2016). The z-scores were converted to p-values and corrected using the method of Benjamini–Hochberg. The statistically significant TFs, and receptors were considered as the reporter biomolecules. The corrected p-value threshold of 0.05 was used to determine statistical significance. Reporter TFs and receptors were examined in GeneCards: The Human Gene Database (Stelzer et al., 2016).
Overrepresentation analyses
Overrepresentation analyses of DEGs and reporter receptors were performed using ConsensusPathDB database (Kamburov et al., 2013). The significantly enriched BPs and molecular pathways were identified using the Gene Ontology (GO) annotation (The Gene Ontology Consortium et al., 2000) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000) database as annotation sources, respectively. The p-values were determined by Fisher's exact test and FDR was applied to control p-values. An adjusted p-value threshold of 0.05 (adjusted-p < 0.05) was used to determine the statistical significance in overrepresentation analysis.
Results
DEGs and overrepresentation analysis
Comparative analysis of gene expression data from aortas of patients with small AAA, large AAA, AOD, and control aortic specimens was performed to identify DEGs and their expression patterns. Statistical analyses resulted in the identification of thousands of DEGs (Supplementary Table S1) and indicated that their expression patterns were predominantly downregulated (87%) (Fig. 1A). In the large AAA, 53 DEGs were upregulated and 1335 DEGs were downregulated, considering the diseased and control states.

A total of 1876 genes, 115 upregulated and 1761 downregulated, were identified as differentially expressed in the small AAA. In the AOD, 836 genes were significantly upregulated, while 3575 genes were significantly downregulated. A total of 1245 mutual DEGs with similar expression patterns were identified between the three disease states. When the expression patterns were examined, 38 upregulated and 1207 downregulated genes were found to be mutual among all disease states (Fig. 1B, C).
Overrepresentation analyses were performed based on the DEGs identified for three disease states, and the 10 most significantly enriched GO BP terms and KEGG pathways are shown in Figure 2. Pathway enrichment analysis revealed infection and immune system-related pathways as mutually overrepresented among the upregulated DEGs in three disease states. Phospholipase D signaling pathway and platelet activation were selectively upregulated only in AOD. Cancer-related pathways, such as cell adhesion molecules and pathways in cancer, were identified to be selectively induced only in the presence of small AAA. Furthermore, the upregulated genes were found to be mainly involved in immunity-associated processes in three diseased states.
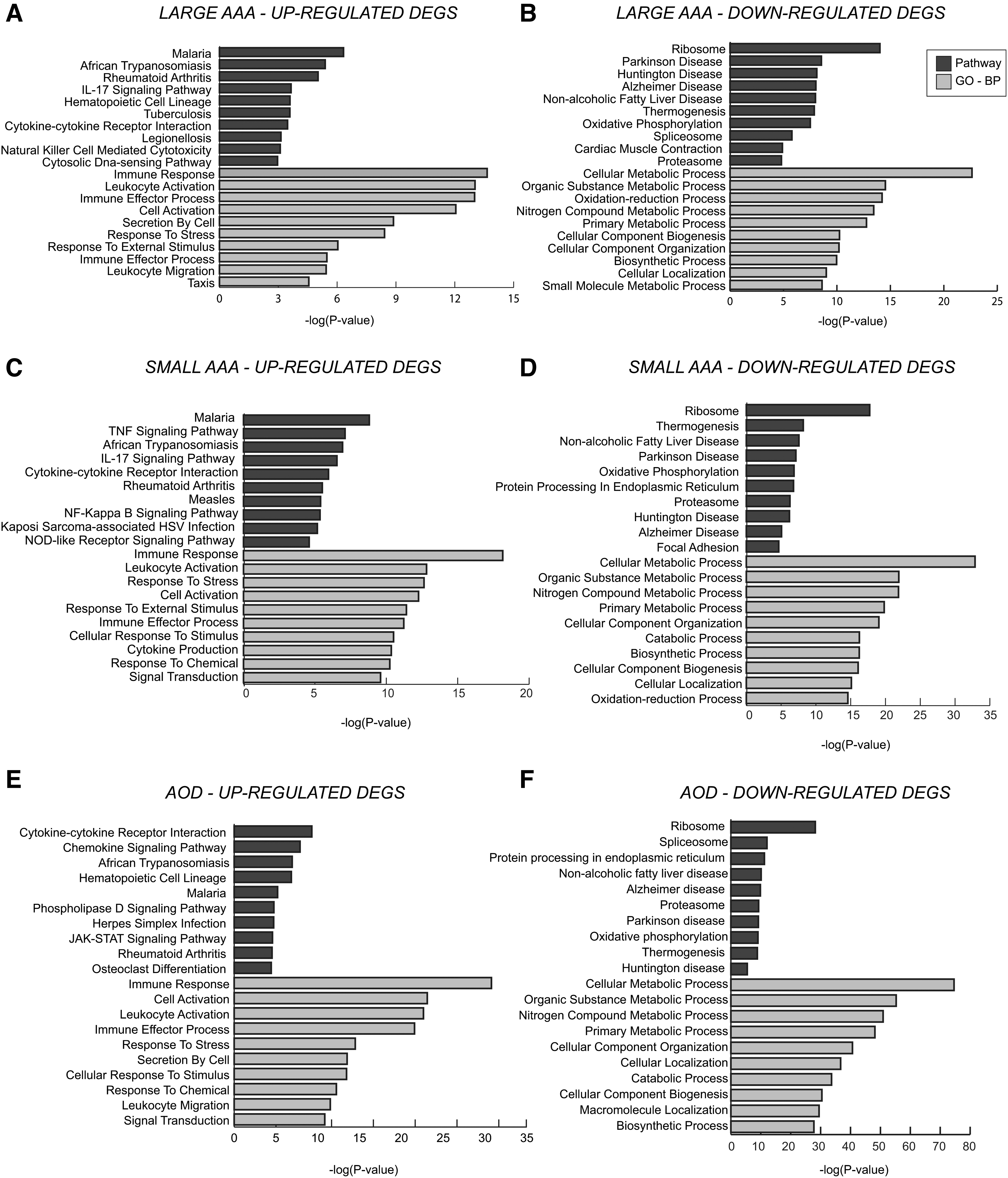
Pathway and GO BP enrichment results of DEGs that were
On the other hand, the pathways related to neurodegenerative disorders, together with oxidative phosphorylation, cardiac muscle contraction, regulation of actin cytoskeleton, thermogenesis, and focal adhesion pathways, were found as the mutual overrepresented pathways of downregulated DEGs in all diseased states.
Central carbon metabolism in cancer, cyclic guanosine monophosphate-protein kinase G (cGMP-PKG) signaling pathway, and vascular smooth muscle contraction were observed among the repressed pathways only in large AAA. Glyoxylate and dicarboxylate metabolism were selectively downregulated only in small AAA. Pathways associated with RNA processing, cell cycle, bacterial invasion epithelial cells, and fatty acid degradation were identified among the repressed pathways only in AOD.
The tricarboxylic acid (TCA) cycle, pyruvate metabolism, and insulin signaling pathways were downregulated in both large AAA and AOD, whereas valine, leucine, and isoleucine degradation, propanoate metabolism, and pathways in cancer were downregulated in both small AAA and AOD. Moreover, the downregulated DEGs were commonly found to be associated with cell cycle, cell death, and myeloid cell homeostasis processes in the three disease states.
Hub proteins
PPI networks were reconstructed around proteins encoded by DEGs and their first neighbors through their physical interactions. In three cases, PPI subnetworks were individually constructed around proteins encoded by upregulated and downregulated DEGs. For large AAA, the upregulated PPI subnetwork consisted of 130 proteins (i.e., 53 upregulated proteins and their first neighbors) and 112 links (i.e., physical PPIs between these proteins), while downregulated PPI subnetwork consisted of 3798 proteins (i.e., 1335 downregulated proteins and their first neighbors) and 7767 links. In small AAA, the upregulated PPI subnetwork was composed of 466 interactions between 470 proteins (i.e., 115 upregulated proteins and their first neighbors), while the downregulated PPI subnetwork consisted of 10,575 interactions among 4596 proteins (i.e., 1761 downregulated proteins and their first neighbors).
The upregulated PPI network of AOD comprised 1975 proteins (i.e., 836 upregulated proteins and their first neighbors) and 2683 interactions, while the downregulated PPI subnetwork of AOD comprised 6278 proteins (i.e., 3575 downregulated proteins and their first neighbors) and 19,226 interactions (Supplementary Fig. S1). Topological analyses of the subnetworks were performed to identify the highly interconnected proteins, that is, hub proteins, which may play an important role in development and progression of the disease.
The 10 proteins with the highest scores of degree and betweenness centrality were determined to be hub proteins and those that were also encoded by DEGs were further analyzed (Table 1). A total of 16, 9, and 14 hub proteins were found to be specific for large AAA, small AAA, and AOD, respectively (Supplementary Table S2). Comparative analysis of hub proteins revealed that VCP is the mutual protein in three cases. ILF3, OGT, COPS5, HNRNPU, and PPP2CA were the mutual hub proteins for both large AAA and small AAA. OGT and ILF3 were upregulated, while COPS5, HNRNPU, and PPP2CA were downregulated in both cases. For small AAA and AOD, two proteins (HIF1A and TNFAIP3) encoded by upregulated genes and three proteins (HSP90AA1, BAG3, and HSPA8) encoded by downregulated genes were mutually identified.
Hub Proteins Associated with the Diseases
AAA, abdominal aortic aneurysm; AOD, aortic occlusive disease.
The diagnostic potential of the hub genes was evaluated using PCA based on the gene expression profiles of 22, 20, and 20 hub genes, respectively, in large AAA, small AAA, and AOD, and sensitivity and specificity metrics were determined. The principal components describing the highest variances revealed the high potential of hub genes in discriminating diseased samples from control samples with sensitivity of 93% (large AAA and small AAA) and 100% (AOD), and specificity of 88% (large AAA), 75% (small AAA), and 100% (AOD) (Supplementary Fig. S2).
Reporter metabolites
Reporter metabolites were identified by integrating gene expression data with the genome-scale human metabolic network. A total of 103, 97, and 113 reporter metabolites were found for large AAA, small AAA, and AOD, respectively (Supplementary Table S3). Comparative analysis revealed 10 mutual reporter metabolites in three cases, and a total of 52, 33, and 88 reporter metabolites specific for large AAA, small AAA, and AOD, respectively were found.
Enzymes and intermediates involved in the degradation of branched-chain amino acids (BCAA), including 2-methylbutyryl-CoA, isobutyryl-CoA, isovaleryl-CoA, S-(2-methylbutanoyl)-dihydrolipoamide, S-(2-methylpropanoyl)-dihydrolipoamide, S-(3-methylbutanoyl)-dihydrolipoamide, dihydrolipoamide, metabolites involved in lipid metabolism, such as sphinganine, and phytanoyl-CoA, and succinate that has roles in TCA cycle and various amino acid metabolisms were identified as the mutual reporter metabolites.
The functional enrichment analyses of the reporter metabolites were also performed to enlighten the metabolic activities around reporter metabolites and oxidative phosphorylation: valine, leucine, and isoleucine degradation pathways were commonly observed in three cases (Table 2). The analyses revealed that amino acid metabolism (alanine, aspartate, and glutamate metabolism) and biosynthesis (valine, leucine, and isoleucine biosynthesis), aminoacyl-tRNA biosynthesis, ABC transporters, cyanoamino acid metabolism, and Parkinson's disease pathways were common in both large and small AAA.
Significantly Enriched Metabolic Pathways and Associated Reporter Metabolites for the Diseases
Fatty acid metabolism came into prominence for large AAA and AOD. Biosynthesis of unsaturated fatty acids and propanoate metabolism were specific to large AAA, whereas several metabolic pathways (i.e., purine, glycine, serine, threonine, and sphingolipid metabolism) and biosynthetic pathways (i.e., phenylalanine, tyrosine, and tryptophan biosynthesis) were specific to small AAA. Fatty acid elongation in mitochondria, glyoxylate, dicarboxylate, and pyrimidine metabolism was highlighted as specific pathways to AOD.
Reporter TFs
The reporter TFs around which the most significant transcriptional changes occur were identified by integrating the transcriptional response in disease states with transcriptional regulatory network using the reporter features algorithm. Disease-specific and common reporter TFs were determined based on the comparative analyses. For each disease state, a total of 14 reporter TFs were identified, which co-occurred in three cases (Supplementary Table S4). Disease-specific reporter TFs were not observed.
Sex-biased genes, including the androgen receptor gene AR, the estrogen receptor gene ESR1, FOXA1 involved in ESR1-mediated transcription, PRDM14 required for the maintenance of embryonic stem cell identity, a set of regulators involved in immunity and inflammation, such as ETS1, which controls the expression of cytokine and chemokine genes (Russell and Garrett-Sinha, 2010), FOXP3, which is crucial for the suppressive activities of regulatory T cells (Li et al., 2015), GATA1-2-3, which are regulators of hematopoietic stem cells and their derivatives (Pikkarainen et al., 2004), YBX1, which is associated with inflammation and stress response (Wang et al., 2019), HIF1A, which regulates adaptive response to hypoxia, E2F4, which is involved in cell cycle regulation, MYC, which is an essential regulator of vascular smooth muscle cell proliferation (de Nigris et al., 2003), and TFAP2C involved in c-Myc-dependent apoptosis (Wang et al., 2020) were identified as reporter TFs in three cases.
Reporter signaling elements: receptors
The reporter receptors of large AAA, small AAA, and AOD were determined using a receptor-protein interaction network. A total of 81, 70, and 17 proteins (Supplementary Table S5) were identified as reporter receptors for large AAA, small AAA, and AOD, respectively. In three cases, no mutual reporter receptor could be found. In addition, 48 reporter receptors were mutual in small and large AAA (Supplementary Table S5). Receptors that play a role in cell cycle and cell death were observed in both large and small AAA (Supplementary Table S6).
Moreover, SV2A, which plays a role in controlling regulated secretion in neural and endocrine cells in both small and large AAA, GPR3, a G protein-coupled receptor with a potential role in modulating a number of brain functions in large AAA, and LRRK2, whose mutations were associated with Parkinson disease in small AAA came into prominence among the top 10 reporter receptors. ABCA4 encoding an ABC transporter, ATP4A involved in ATP hydrolysis, ATAD2, which may be a transcriptional coactivator of the nuclear receptor ESR1 required to induce the expression of a subset of estradiol target genes, ATP8, which was previously associated with cardiomyopathy, and two receptors (CD2 and CLCN3) related to immunity were highlighted among the top 10 reporter receptors identified in the presence of AOD.
Discussion
Among CVDs, AAA and AOD share similar risk factors and mechanisms of pathogenesis; however, recent studies suggest that their pathologies also display divergence. The elucidation of the diversity of molecular mechanisms would increase our understanding of pathogenesis of each disease and provide valuable information for predictive biomarker discovery and development.
Despite the molecular studies on AAA and AOD (Biros et al., 2015; Ciborowski et al., 2012), the mechanisms underlying these diseases are still not fully understood. Currently, early diagnosis of AAA is generally attained by incidental imaging or screening programs (Golledge, 2019), and the diagnosis largely depends on the physician experience and the selected method. Due to the lack of robust diagnostic and prognostic biomarkers, alternative diagnostic methodologies would be invaluable to overcome the difficulties of screening-based methods (Moris et al., 2014; Zhang et al., 2015). Therefore, in this study, an integrative analysis of genome-wide transcriptome data with comprehensive human biological networks was performed to elucidate the mechanisms underlying the pathogenesis of AAA and AOD and to uncover biomolecules that play key roles in the mechanisms of these diseases.
Specifically, we identified the DEGs and associated molecular pathways and BPs in each disease. Using a holistic approach, these data were linked to comprehensive human biological networks (i.e., PPI, transcriptional regulatory, and metabolic networks) to identify molecular signatures of salient mechanisms of AAA and AOD. PPI networks were reconstructed for all disease states and topological analyses of these networks revealed hub proteins that may play central roles in disease progression. In addition, integrative analysis of gene expression data with a genome-scale metabolic model and a transcriptional regulatory network identified reporter biomolecules (metabolites, receptors, and TFs, respectively) that warrant future development as potential prognostic biomarkers and putative therapeutic targets.
The independent and integrative analyses indicated that key biological pathways and processes were downregulated in the presence of AAA and AOD. Infection-associated signaling pathways, the immune response, and oxidative phosphorylation were among the commonly affected pathways in all diseases (Fig. 3). In addition, alterations and differences were observed in BPs and metabolic pathways related to lipid metabolism, valine, leucine, and isoleucine metabolism, and neurodegenerative diseases (i.e., Alzheimer's, Parkinson's, and Huntington's disease), and sex differences came into prominence in the presence of AAA and AOD (Fig. 3).
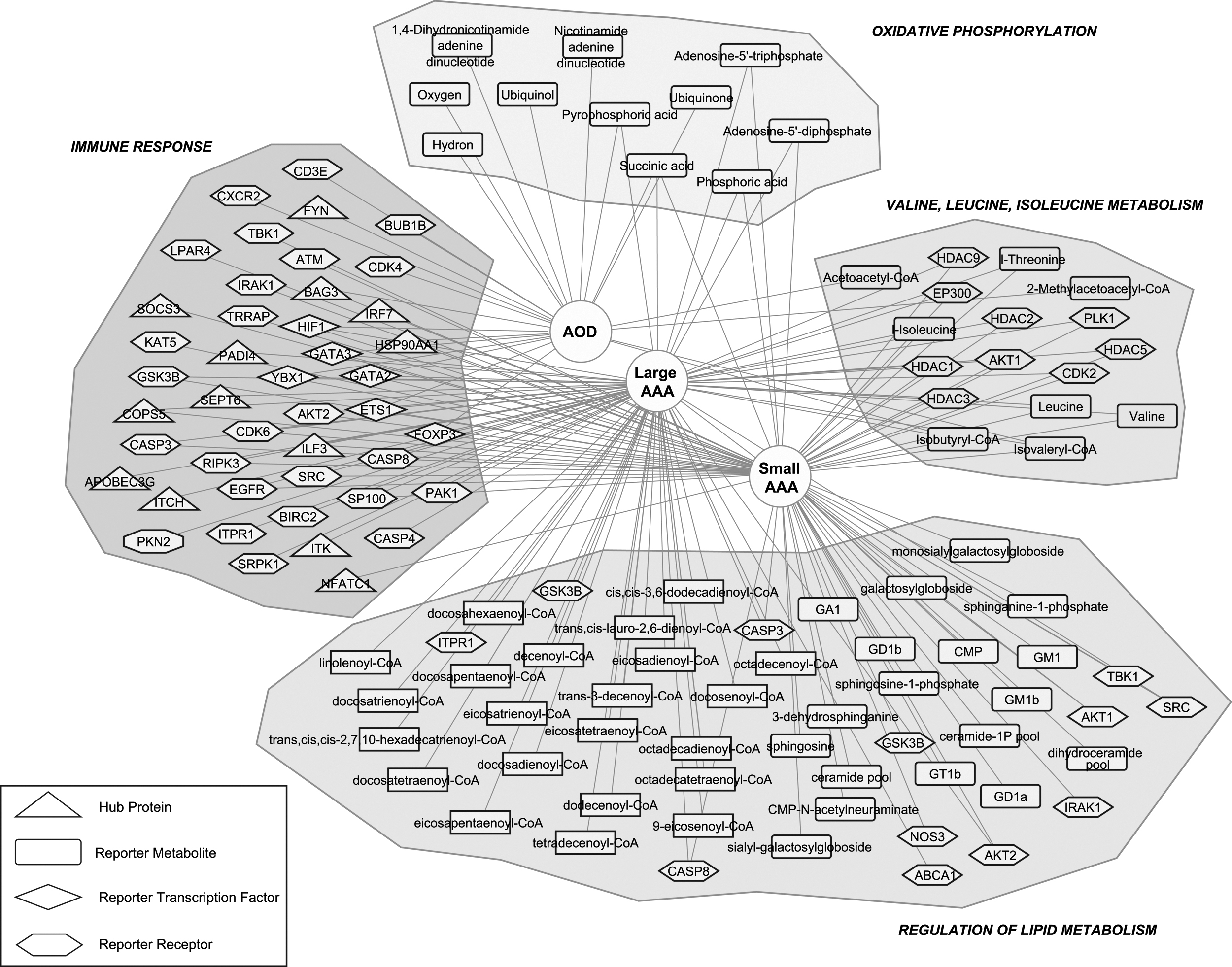
A conceptional summary of the biomolecules identified as molecular signatures in small AAA, large AAA, and AOD.
Immune response
Both atherosclerosis and AAA are arterial wall diseases characterized by severe inflammation; however, the roles of immune-mediated mechanisms are not same in AOD and AAA. Although the critical roles of inflammation and immunity have been accepted for AOD, the nature of immune-mediated mechanisms still need to be addressed for AAA (Peshkova et al., 2016), and our results revealed similarities and alterations in immune system-associated pathways in both diseases.
The comparative analysis of DEGs indicated immune response-associated processes and pathways, together with pathways of infectious diseases as mutually overrepresented in small AAA, large AAA, and AOD. Furthermore, analysis of PPI networks revealed hub proteins that play roles in immunity, inflammation, and infectivity. Among these proteins, APOBEC3G, ITK, PADI4, SEPT6, and ITCH were selectively identified in large AAA; IRF7, NFATC1, and SOCS3 were found to be specific to small AAA; and FYN was identified only in AOD.
Besides disease-specific proteins, two proteins encoded by immune response-related genes, ILF3 and COPS5, were identified commonly in large and small AAA. ILF3 and COPS5 have been shown to be associated with various CVDs; however, their roles in AAA have not been elucidated yet. ILF3, which is required for T cell expression of interleukin 2, has been suggested to be involved in lipid profile-dependent atherosclerotic calcification in mice (Xie et al., 2021). The accumulation of ILF3 in the necrotic core of coronary plaque and the association of ILF3 polymorphism with myocardial infarction in individuals with low levels of low density lipoprotein (LDL) cholesterol (Yoshida et al., 2011) may suggest a possible function of ILF3 in AAA pathogenesis.
COPS5 has been reported to play a role in the development of atherosclerosis (Milic et al., 2019), which in most cases co-exists with AAA (Soto et al., 2017). In addition, in an atherogenic mice model, COPS5 has been shown to function in reducing atherogenesis by blocking inflammatory signaling in myeloid cells (Asare et al., 2017). Moreover, HSP90AA1 involved in the activation of innate immune system, and BAG3, whose expression induces by viral infection (Rosati et al., 2011), were determined as common hub proteins of small AAA and AOD. Loss of BAG3 functions led to the development of dilated cardiomyopathy (Knezevic et al., 2015), and the upregulation of BAG3 was reported to improve cardiac function and reduce cardiomyocyte-related deaths in mice (Mizushima and Sadoshima, 2017).
Possible protection of HSP90AA1 recovery against myocardial ischemia/reperfusion injury has also been suggested based on its regulation by miR-1, which is involved in myocardial ischemia/reperfusion injury (Zhu et al., 2016). In addition to altered expression levels of genes associated with immunity, we identified hub proteins that play roles in immune response in all diseased states.
The integration of transcriptome and regulome revealed FOXP3, ETS1, YBX1, HIF1, GATA2, and GATA3 that are involved in immune response and inflammation as common TFs in the studied diseases. FOXP3 expression has been shown to be altered in inflammatory diseases (Gorzin et al., 2017) and AAA (Moris et al., 2014). Defects in T cell and B cell development and in vascular inflammation were observed in ETS1-deficient mice and a possible role of ETS1 in the etiology of congenital heart disease has been suggested (Gao et al., 2010). YBX1, a member of Y-box protein family that is associated with inflammation and stress responses, had a protective role in oxidative stress-induced cardiac injury (Wang et al., 2019) and has been suggested as a potential target for myocardial fibrosis (Lin et al., 2019).
HIF1A has an important role in diabetes. In a mouse model, loss of function of hypoxia-inducible factor 1 (HIF-1) in diabetic pregnancies resulted in alterations in immune system-related processes and inflammatory responses. The reduced protein expression levels of HIF-1 as a result of exposure to maternal diabetes were reported to be associated with impaired cardiac function and are considered one of the key factors in fatal reprogramming of CVD in adulthood (Cerychova et al., 2018). The dysregulation of GATA2 has been associated with immunodeficiency syndromes, and genome-wide transcriptional analyses provided evidence for repression of cardiac development-related genes by binding GATA2 to regulatory regions.
GATA3 had functions in T cell development (Tindemans et al., 2014). The identification of FOXP3, ETS1, YBX1, HIF1, GATA2, and GATA3 as common TFs supported the importance of the regulation of inflammatory and immune responses in the studied diseases.
Owing to the key roles of receptors in signal transduction, reporter receptors provided valuable information on the biological alterations in the cells due to disease pathogenesis. The identified reporter receptors were found to be enriched with various infectious disease pathways in both large AAA and small AAA. T cell receptor signaling pathway was also identified as the overrepresented pathways associated with receptors in large AAA and AOD. Receptors involved in immune system development, cytokine production, and leukocyte activation were observed in the presence of small AAA and large AAA.
The functional impairment of regulatory T cells has been previously reported in various CVDs, including AAA, atherosclerosis, hypertension, myocardial infarction, and heart failure, and inflammation, contributed to the progression of these immune-response associated diseases (Meng et al., 2016). Previous studies also indicated an important role of immune responses in the pathogenesis of AAA (Biros et al., 2015), and in the regulation of inflammation in atherosclerosis (Roy et al., 2021). Therefore, the identified biomolecules could be assessed as prognostic markers for AAA and AOD.
Oxidative phosphorylation
Comparative analysis of DEGs in the presence of large AAA, small AAA, and AOD resulted in the identification of commonly downregulated genes associated with oxidative phosphorylation. Moreover, the pathway enrichment analysis of reporter metabolites indicated that oxidative phosphorylation has been highlighted with four, five, and eight metabolites in small AAA, large AAA, and AOD, respectively (Table 2).
Disorders of cardiac mitochondrial oxidative phosphorylation have been reported to play an important role in the development of various CVDs. A shift from oxidative phosphorylation to glycolysis in the heart of patients with coronary artery disease (Ait-Aissa et al., 2019), impaired mitochondrial oxidative phosphorylation capacity, elevated mitochondrial reactive oxygen species (ROS) in the heart of cats with hypertrophic cardiomyopathy (Christiansen et al., 2015), and a link between impaired oxidative phosphorylation and coronary atherosclerosis (Nakajima et al., 2019) have been reported previously.
Moreover, the observation of a preferential increase in mitochondrial DNA damage in vascular smooth muscle cells and endothelial cells in response to exposure to ROS provided evidence for the impairment of endothelial function and induction of vascular smooth muscle cell apoptosis, leading to the development of atherosclerosis (Ballinger et al., 2000; Madamanchi and Runge, 2007). Our findings indicated that the genes playing roles in vascular smooth muscle contraction were downregulated in both small AAA, and AOD. Therefore, the lower expression of these genes might also provide further support for the development of AAA and AOD due to impaired oxidative phosphorylation.
Regulation of lipid metabolism
The independent analysis of DEGs in the presence of studied diseases indicated repression of genes involved in fatty acid degradation pathway in AOD. Receptors involved in lipid and atherosclerosis pathway both in small AAA and large AAA, and metabolites involved in fatty acid metabolism and fatty acid elongation in AOD were identified. Furthermore, the analysis of reporter metabolites on AAA highlighted the changes in lipid metabolism depending on the size of AAA. Thirty-one metabolites involved in the β-oxidation of fatty acids, and 23 metabolites involved in the carnitine shuttle were found to be specific for large AAA. Carnitine plays an essential role in the transport of long-chain fatty acids into mitochondria for β-oxidation, and carnitine deficiency has been reported to be significantly associated with AAA (Arsenian, 1997; Ciborowski et al., 2012).
In contrast, when the small AAA-specific reporter metabolites were investigated, 18 metabolites that have functions in sphingolipid metabolism came to the fore. There is significant evidence that disruption of the balance between the two main sphingolipids, ceramide and sphingosine-1-phosphate, leads to the induction of apoptosis, which is involved in the pathogenesis of CVDs (Borodzicz et al., 2015). Moreover, ceramides that accumulate in diseased tissues are highly pathogenic, and in cardiometabolic diseases, including coronary artery disease, diabetes mellitus, cardiomyopathy, and heart failure, and ceramides are responsible for much of the tissue damage (Choi et al., 2021).
Dysregulation of lipid metabolism was observed in both small and large AAA. Moreover, as the size of the aneurysm increased, a shift from sphingolipid metabolism to the β-oxidation of fatty acids and the carnitine shuttle was noted. Our results suggest that these reporter metabolites can be used to discriminate between small and large AAA, and thus to assess the stage of AAA. Furthermore, the identified metabolites involved in sphingolipid metabolism could be considered system-level biomarkers for early detection of AAA and used to assess the risk of rupture; however, further clinical investigation is required to confirm these findings.
Valine, leucine, and isoleucine metabolism
Valine, leucine, and isoleucine metabolism has come to the fore at mRNA, metabolite, and protein levels. The genes involved in valine, leucine, and isoleucine degradation pathway were downregulated in small AAA and AOD. Moreover, the pathway enrichment analysis of reporter metabolites revealed valine, leucine, and isoleucine degradation pathway commonly in three disease states. Seven of the 10 common reporter metabolites were enzymes and intermediates involved in the degradation of BCAA.
Abnormal BCAA metabolism has previously been shown to be associated with coronary diseases (Huang et al., 2011). Cardiac BCAA accumulation has been observed in dilated cardiomyopathy (Adeva-Andany et al., 2017; Uddin et al., 2019), and defects in BCAA catabolism have been associated with metabolic syndrome, such as insulin resistance, obesity, and diabetes mellitus (Adeva-Andany et al., 2017; Huang et al., 2011). In addition, reducing cardiac BCAA metabolites by accelerating of BCAA oxidation has been suggested as a potential treatment for heart failure (Uddin et al., 2019).
A total of 26 DEGS were found to be involved in valine, leucine, and isoleucine metabolism in three disease states, and the interactions of reporter receptors with proteins encoded by these DEGs were investigated. Although reporter receptors for AOD were not found to be associated with this metabolism, 11 receptors were highlighted in small or large AAA. Among these receptors, AKT1, one of the three isoforms of AKT, five histone deacetylases (HDAC1, HDAC2, HDAC3, HDAC5, and HDAC9), EP300, PLK1, and CDK2 were determined in both small and large AAA. AKT isoforms have associated aneurysm formation (Yu et al., 2015), and a significant increase in AKT phosphorylation has been reported in male compared with female mice during AAA progression (Ghosh et al., 2014).
Induction of histone deacetylases in human AAA and limitation of AAA progression with histone deacetylase inhibitors in mouse model have been reported previously (Galán et al., 2016). EP300, which functions as a histone acetyltransferase, could be a potential target for inherited AAA because some patients carrying EP300 mutations have ascending aorta (Costain et al., 2018) or congenital aortic aneurysm (Fergelot et al., 2016). PLK1, which is involved in cardiovascular homeostasis, has been associated with elevated risk of aneurysm and aortic rupture (De Cárcer et al., 2017). Although cyclin-dependent kinase inhibitory drugs have been proposed as potential agents against inflammatory diseases (Leitch et al., 2009), CDK2 has not been associated with AAA.
BMPR2 (BMP type II receptor) has been identified in large AAA. Mutations in BMP signaling genes resulted in vascular dysfunction and various diseases, including pulmonary arterial hypertension, which can lead to pulmonary artery aneurysm (Cai et al., 2012; Nuche et al., 2020). TBK1, which plays an important role in controlling energy metabolism and attenuating of inflammation (Zhao et al., 2018), was identified in small AAA. Our study proposed the association of AAA with CDK2, BMPR2, and TBK1 receptors for the first time, and further efforts for clinical validation are required.
Sex differences
The risks, incidences, and outcomes of CVDs differ depending on gender, and therefore, the consideration of sex differences becomes crucial for the prevention, diagnosis, and treatment of these diseases (Gao et al., 2019). In the case of AAA, the prevalence in men is around four to six times higher when compared to women, and male sex has been considered among the risk factors for the development and progression of AAA (Gao et al., 2019; Huang et al., 2016). In addition, for women, it was reported that the risk of developing a CVD is lower until menopause compared with men of the same age (Puzianowska-Kuznicka, 2012). However, after menopause, the risk of AAA increases due to a decrease in protection by hormones (Gao et al., 2019).
The effects of sex differences on the occurrence and progression of AAA and AOD were mainly observed at transcriptional regulation level. Four common sex-biased TFs (AR, ESR1, FOXA1, and PRDM14) have come to the fore. AR signal transduction and/or deficiency have previously been linked to CVDs, including atherosclerosis, and AAA. Increased lipid accumulation due to androgen deficiency enhances atherosclerosis, and androgen signaling has a considerable effect on the development and progression of the AAA. A mouse model showed that AR could modulate the formation of AAA by altering inflammation to maintain aortic wall integrity (Huang et al., 2016).
Moreover, ESR1, FOXA1, and PRDM14 have been associated with various CVDs, but their associations with AAA and AOD have not yet been identified. ESR1 is an estrogen receptor gene and FOXA1 is a key determinant of estrogen receptor binding in breast cancer cells. However, its expression also resulted in alterations in estrogen receptor binding and function in non-breast cancer cells (Hurtado et al., 2011). These findings were consistent with previous studies suggesting an important role for estrogen receptors in cardiomyocyte homeostasis and cardioprotection. PRDM14 has previously been reported as a regulator of sex-biased genes during mouse cardiac development and in the adult heart (Deegan et al., 2019). Therefore, the identified sex-biased TFs could be evaluated as potential markers for screening and therapeutic purposes in AAA and AOD.
Conclusions
Among CVDs, AAA and AOD are of great public health importance because of the high mortality rate in the elderly population. A better understanding of metabolic processes and pathways underlying the pathogenesis of AAA and AOD and the identification of molecular biomarkers are vital for the development of novel diagnostic and therapeutic strategies. We have reported the similarities and differences in the re-organization of the transcriptomic response as a result of the progression of AAA and AOD, and identified novel proteomic, metabolic, and regulatory molecules as potential biomarkers and putative therapeutic targets for further investigation. However, future studies are required for experimental and clinical validation of findings obtained here to provide novel insights to design diagnostic markers and treatment strategies for AAA and AOD.
Footnotes
Authors' Contributions
C.K. and K.Y.A. conceived the study. D.C. and M.K. carried out the analyses. M.K., C.K., and K.Y.A. interpreted the results. D.C., M.K., and C.K. wrote the article. C.K. and K.Y.A. finalized the article with critical reading. All authors read, revised, and approved the final version of the article.
Acknowledgments
The scholarships under the BIDEB 2211/C National PhD Scholarship Program by The Scientific and Technological Research Council of Turkey (TÜBİTAK) and under the YÖK 100/2000 Doctoral Fellowship Program by the Council of Higher Education (YÖK) provided to M.K. are greatly acknowledged.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.