
Abstract
The majority of processes that occur in daily cell life are modulated by hundreds to thousands of dynamic protein–protein interactions (PPI). The resulting protein complexes constitute a tangled network that, with its continuous remodeling, builds up highly organized functional units. Thus, defining the dynamic interactome of one or more proteins allows determining the full range of biological activities these proteins are capable of. This conceptual approach is poised to gain further traction and significance in the current postgenomic era wherein the treatment of severe diseases needs to be tackled at both genomic and PPI levels. This also holds true for COVID-19, a multisystemic disease affecting biological networks across the biological hierarchy from genome to proteome to metabolome. In this overarching context and the current historical moment of the COVID-19 pandemic where systems biology increasingly comes to the fore, cross-linking mass spectrometry (XL-MS) has become highly relevant, emerging as a powerful tool for PPI discovery and characterization. This expert review highlights the advanced XL-MS approaches that provide in vivo insights into the three-dimensional protein complexes, overcoming the static nature of common interactomics data and embracing the dynamics of the cell proteome landscape. Many XL-MS applications based on the use of diverse cross-linkers, MS detection methods, and predictive bioinformatic tools for single proteins or proteome-wide interactions were shown. We conclude with a future outlook on XL-MS applications in the field of structural proteomics and ways to sustain the remarkable flexibility of XL-MS for dynamic interactomics and structural studies in systems biology and planetary health.
Introduction
Protein–protein interactions (PPI) regulate every aspect of cell life, including immune recognition, communication across tissue districts, synaptogenesis, and even cell functioning or death by activation of autophagy or apoptosis pathways (Basu et al, 2021; Gonzalez Melo et al, 2021b; Manganelli et al, 2021). A consistent organization of PPI networks plays a central role in terms of accurate operation of behavior, governing multicellular organisms through fine regulation mechanisms. Besides PPI, proteins establish multiple connections with other molecules and this lets one suppose the existence of extremely convoluted maps that define the cellular interaction landscapes (Diether and Sauer, 2017). Coherently, at every level considered, the great advantage offered by the omics perspective is to provide specific molecular signatures that mirror the ongoing cellular processes, in both health and disease states (Barigazzi et al, 2020; Costanzo et al, 2022d; Costanzo et al, 2017; Olivier et al, 2019; Santorelli et al, 2021a; Santorelli et al, 2021b).
PPI can be classified as either permanent or transient. Permanent interactions result in the formation of strong protein complexes. For instance, the aggregation of proteins with similar chemical–physical properties identifies a bunch of genes functionally involved in a common pathway. However, most interactions are transient, weak, occurring only for a brief period, and largely influenced by the nature of their molecular composition. That biological strategy increases exponentially the cell capability to control a plethora of intracellular and extracellular phenomena. It is reported that at least one third of soluble proteins exist in multimeric state (McBride et al, 2019), composing multiprotein complexes that modulate inner events, such as DNA replication, DNA repair, and gene expression (Gontier et al, 2021; Rulten and Grundy, 2017), as well as faraway processes, including cell adhesion and motility, vesicular trafficking, and signaling (Chan et al, 2014; Lopes-da-Silva et al, 2019; Raimondo et al, 2018).
Proteins spontaneously associate and disassociate, according to cellular needs, thus ensuring the normal cell life. However, when this knotty balance between assembly and disassembly is hampered, the effects can impact on organs and tissue functionality. Moreover, aberrations in protein complex organization have been widely documented in the past decades. Protein aggregation has been shown to be associated with diverse amyloid, age-related, and neurodegenerative diseases (Burrinha et al, 2021; Chuang et al, 2018; Farris et al, 2021). Furthermore, mutations at dimer interface, post-translational modifications (PTMs), addition/loss, and insert/deletion of specific domains interfere with protein capabilities to interact between them. These mechanisms underlie several genetic disorders and oncogenic diseases (He et al, 2021; Makita et al, 2012; Shen et al, 2017; Singh et al, 2019).
Thus, both the identification of simple PPI in cells and the dissection of intricate functional networks (among distant districts or between protein of different organisms or dependent on drugs or other treatments) efficiently disclose the complex functioning of distinct, but even correlated biological processes, improving the knowledge of cellular normal functions as well as the causal basis of different disorders (Costanzo et al, 2021b; Gonzalez Melo et al, 2021a). In fact, some disease mechanisms alter existing PPI or functional networks, by removing some protein domains indispensable for the network connection (Mukherjee et al, 2022).
For instance, such conditions may be targeted for the development of PPI-based therapies or similar treatments in the context of personalized medicine. As an example comes the brilliant work of Gordon et al (2020) in the context of the current COVID-19 pandemic. These authors, through the expression of 26 of the 29 SARS-CoV-2 proteins in human cells and using an affinity-purification mass spectrometry (MS) approach, obtained the identification of the human proteins physically interacting with SARS-CoV-2 proteins, including a total of 332 highly confident PPI between human and coronavirus proteins. Beyond their discovery, they found several human proteins or host factors targetable by drugs or compounds with subsequent antiviral activity (Gordon et al, 2020).
Establishing the structure of an interactome is directly correlated with the discovery of its function within the cells, considering that if two proteins are identified as reciprocal interactors, they are supposed to be functionally associated (Guala et al, 2020; Huynen et al, 2000). Functional association networks are important because they represent a snapshot of simultaneous cellular events; thus, there is the need to consider a load of protein interaction evidence, deriving from diverse sources that may consider not only physical interactions.
In particular, due to the fact that proteins can associate in disparate manners, data integration from diverse sources is mandatory to build an accurate functional association network, increasing the overall network quality (Jansen et al, 2003; Xing and Dunson, 2011). Several databases that take into account the diverse orders of evidence (i.e., prior knowledge from curated database, computational prediction of interactions, experimentally proven interactions) are available for assisting researchers especially in proteome research, such as STRING (Drongitis et al, 2022; Szklarczyk et al, 2021), IntAct (del Toro et al, 2022), BioGRID (Oughtred et al, 2021), and many others.
To dissect the intricacy of the cellular connections, mass spectrometry coupled with protein cross-linking (XL-MS) appears to be one of the most promising strategies to identify and map PPI, capturing the dynamics of interactomes from small scale to large scale (Fig. 1). In combination with other techniques able to define the structure of molecules, XL-MS places itself as a valid complementary tool to combine with classical structural biology methodologies, such as X-ray crystallography, cryoelectron microscopy (cryo-EM), and nuclear magnetic resonance (NMR) spectroscopy (Braitbard et al, 2019; García-Nafría and Tate, 2021; Musielak et al, 2020; Roviello et al, 2014).

A conceptual diagram that shows the flux of work and information obtainable by XL-MS through the analysis of the interactomes. XL-MS, cross-linking mass spectrometry.
In this review, we summarize the current knowledge behind the XL-MS technology (i.e., cross-linking reagents, MS acquisition methods, and data analysis programs), reporting the main advantages of this technique and providing an overview of the latest tools and workflows developed for proteome research. Some conceptual caveats related to XL-MS were also handled to highlight the flexibility of this technique at every level of sample complexity, from in vitro purified proteins to the system-wide proteome analysis, especially in the field of structural proteomics.
In fact, in this study, we support the potentialities of XL-MS from the low-scale analysis to the proteome-wide identification of functional connections in tissues, living samples, or whole organisms.
XL-MS: Principles and Methodology
In the field of three-dimensional (3D) protein organization, XL-MS represents a valid strategy capable of achieving interesting results, first allowing to determine the composition and distribution of protein units in well-established macromolecular complexes that the conventional experimental procedures may fail to characterize. Second, it can also provide in vivo insights that build upon the current static interactomics data, to include dynamic information of PPI changes, in the native status or in response to specific stimuli from the environment. XL-MS becomes immensely powerful when applied in a global-scale manner, driving to the enlightenment of novel binding partners that would otherwise not be detected. Additionally, beside the interprotein interactions, XL-MS provides for each component of a protein complex also information related to the intraprotein connections, which further allow to elucidate the structural architecture of multi-subunit aggregates (Fürsch et al, 2021; Vallat et al, 2018; Zhong et al, 2020).
A schematic example for a general XL-MS workflow is shown in Figure 2. Single protein complexes or whole proteomes are first reacted with cross-linking reagents to hold the existing interactions through the formation of covalent bonds between proximal domains of interacting partners. The cross-linked proteins may be separated by electrophoresis and in situ digested, or directly in-solution hydrolyzed. Several chromatographic techniques or new fractionation methods have been successfully employed for separating cross-linked peptides, thus enriching the cross-linked peptides-containing fractions and extending the coverage of the proteome during the following MS analysis, especially for large-scale studies (Jiao et al, 2022; Wu et al, 2021). In fact, the cross-linked peptides (X-linked) are hopefully separated from the unmodified (non-X-linked) ones and analyzed through liquid chromatography–tandem mass spectrometry (LC-MS/MS).

Schematic of a generic XL-MS workflow. The arrows indicate the subsequential steps that follow from the cross-linking reaction to the discovery and definition of dynamic PPI and interaction networks. Some pieces of this figure were adapted from SciDraw (https://www.scidraw.io). PPI, protein–protein interaction.
The acquired data are searched against dedicated databases and elaborated by specific bioinformatics tools to identify and quantify cross-linked peptides sequences and the cross-modified sites. Finally, the localization of the cross-linked sites and their distribution across the proteins are used to build up XL-maps, ranging from the definition of single protein structures to the de novo predictions of interproteins connections. Such strategy allows the definition of simpler PPI and more complex PPI network within the cells, opening the view to a large spectrum of possibilities, which include the definition of the structural dynamics of the connections and the modulation of their possible conformations through diverse regulative mechanisms. Furthermore, the application of methods for integrative data assembly that take into account information obtained with other structural tools may provide details of ensembles through advanced molecular modeling approaches (Czaplewski et al, 2021).
A global overview of the most common tools utilized for general XL-MS workflows is shown in Figure 3. Several types of cross-linkers, as well as the MS fragmentation and acquisition methods, the quantitative strategies for XL-MS (QXL-MS), and the computational search tools available are reported and described in detail in the following sections of this article.
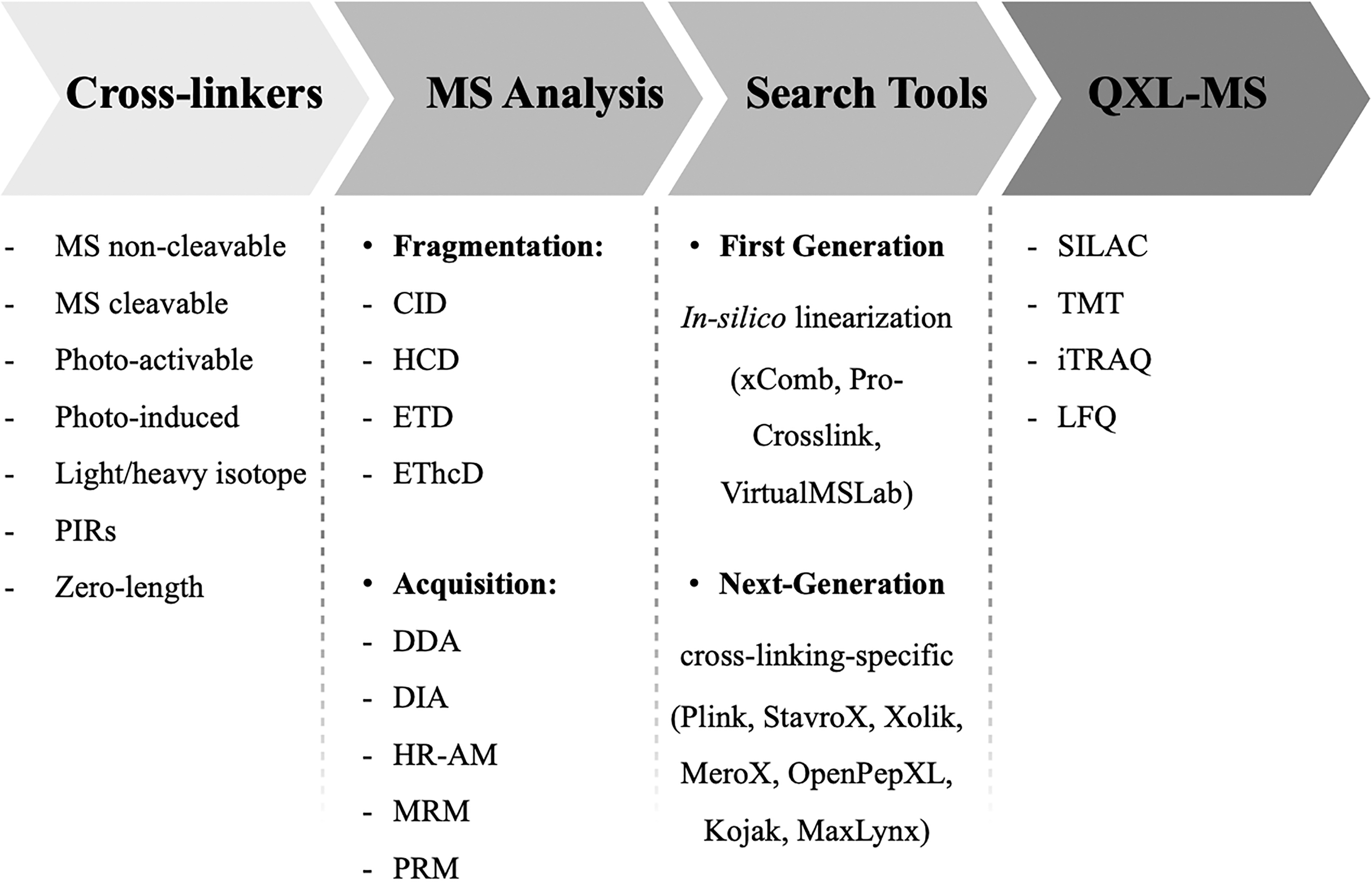
Prospect of the several analytical and computational approaches and tools that can assist an experimental workflow based on XL-MS.
Cross-linkers: types and chemical functionality
In case of XL-MS studies, primary amines are excellent targets for cross-linking due to their reactivity and relatively high abundance at the surface of proteins. Thus, standard cross-linking reagents, such as disuccinimidyl suberate, bis-(sulfosuccinimidyl) suberate, bis-(sulfosuccinimidyl) glutarate, and diethyl suberthioimidate, are typically used for their reactivity with lysine residues and N-termini of proteins, and to a lesser extent with serine, threonine, and tyrosine (Lauber and Reilly, 2010; Steigenberger et al, 2020). Generally, a two- or threefold molar excess of cross-linker over the amount of proteins results in sufficient conjugation ratio between proximal groups. The establishment of the best working ratio between cross-linker and protein molecules is a crucial step.
Indeed, after the cross-linking and digestion phases, four reaction products with specific links combination could be obtained, as shown in Figure 4: (i) intramolecular bonds, occurring within the same molecule, are interesting in case of 3D single protein studies; (ii) intermolecular bonds, the most informative in the case of PPI analysis, identify what parts of different proteins form the interaction interface; (iii) monolinker bonds, peptides modified with a hydrolyzed cross-linker portion; not all cross-linking reagents will hydrolyze; moreover, such products will also result from a quenching step to deactivate the remaining reactive group in reagents that hydrolyze; (iv) loop-linker bonds, intrapeptide links generated by the reaction between two close amino acids in the same peptide sequence. These last two conformations are generally considered side effects of an over-numerical presence of cross-linker molecules compared with protein content.
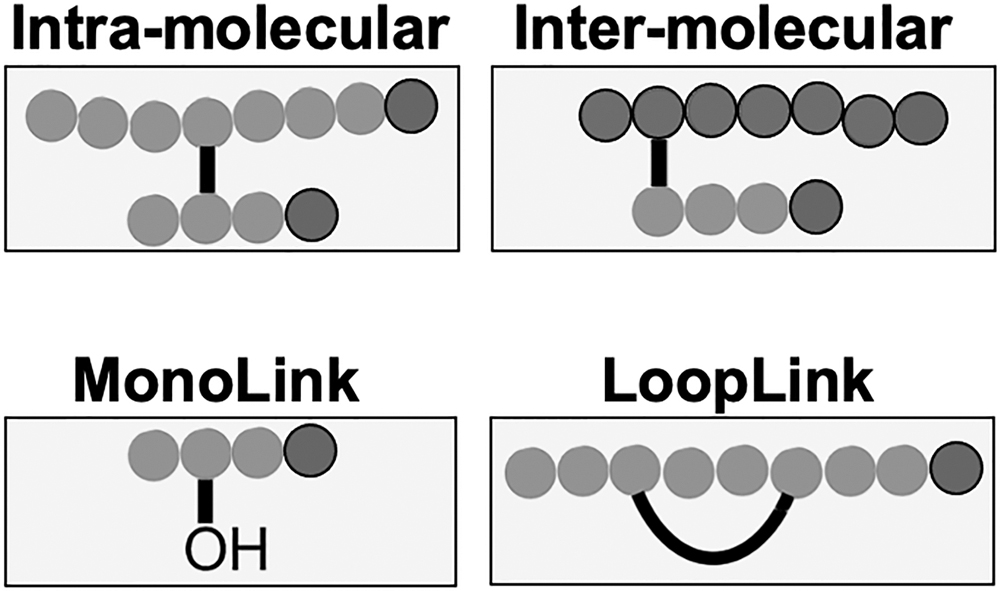
Possible combinations of peptidic connections obtainable using cross-linking reagents. The nomenclature of common products of chemical cross-linking reactions is shown in figure. Intra- and intermolecular links, which connect two peptides, are examples of type 2 cross-links. The loop link is an intrapeptide link (single peptide) recognized as type 1 cross-link. Finally, the monolink is a dead-end link (single peptide) recognized as type 0 cross-link.
The formation and the identification of intermolecular bonds provide clues regarding the optimization of the proteome to establish a precise topology, revealing the need of a particular function in a given time. However, the intramolecular bond within the same protein molecules is the most frequent type of specific interaction. These links possibly generate homomultimers, owing to the forced co-localization of homomultimers partners and their high concentration within the same near physical space (Bruce, 2012).
This consideration is further endorsed by the fact that the genetic evolution through the selection of advantageous mutations supports the most beneficial protein topological features. Such aspects explain the similar effect of PTMs on protein topology carried out through the modification of some amino acid residues, by changing their size, charge, or hydrophobicity (Bruce, 2012; Santorelli et al, 2020a; Santorelli et al, 2020b). Recently, a detailed protocol was published on how to differentiate intramolecular from intermolecular links in multimeric complexes to study homodimer interfaces (Lima et al, 2018).
The molecules described so far may be classified as non-cleavable cross-linkers. They represent the first chemical cross-linkers generation and the most commonly used in structural MS experiments at the beginning. Their adoption has been fundamental for the 3D configurational determination of small-scale protein networks, where the acquired MS spectra are numerically limited, consequently rendering the data analysis relatively straightforward to manage (Ryl et al, 2020; Slavin and Kalisman, 2018). Conversely, composite peptide mixtures are notoriously difficult to handle; this is particularly true in the case of XL-MS protocols that utilize such reagents for cross-linking.
However, a similar strategy suffers from two main problems. First, the possible reduction of the confidence interval in the identification assignment of cross-linked peptides, consequence of quadratic expansion of the computational search space. The reason for such expansion (also known as the “n-square” problem) lies in exponentially amplifying the theoretical peptide pairing to examine against the database, considering the intact masses of the cross-linker domain and the individual peptides (Liu et al, 2015). The second issue is the unequal fragmentation efficiency of the two linked peptides. Nonetheless, it is largely reported that in case of higher energy collisional dissociation (HCD) MS methods, the formation of product ions from only one of the two constituent peptides is generally preferred (Liu et al, 2015). This impacts on the confident assignment of cross-links sites and residue sequences involved in the bond, once more.
With the aim of overcoming those limitations, a second generation of cross-linkers with MS-cleavable properties were introduced. These novel MS-cleavable reagents, such as disuccinimidyl dibutyric urea, disuccinimidyl sulfoxide (DSSO), and 1,1′-carbonyldiimidazole, present labile bonds that are selectively cleaved during the MS fragmentation (Ihling et al, 2020; Matzinger and Mechtler, 2021; Ser et al, 2019; Tveen-Jensen et al, 2013). Hence, data complexity is considerably reduced considering that MS-cleavable reagents grant the generation of characteristic reporter ions and provide the mass of the individual peptides rather than that of the cross-linked moiety (Stieger et al, 2019). Thus, their adoption allows obtaining more accurate data and abates the misidentification rate during the database searching step, especially in case of complex peptide mixtures, as for whole proteomes. Both non-cleavable and MS-cleavable cross-linkers are suitable for a XL-MS workflow, employing optimized gas-phase fragmentation methods (e.g., CID, HCD, ETD, and EThcD) (Stieger et al, 2019).
Apart from this major distinction, there are other types of linkers classified according to their photochemical reactivity. These include photoactivatable cross-linkers that react with target molecules by exposure to UV light and photo-induced cross-linkers, whose reaction is mediated by high-valent metal–chelate compounds (Henderson and Nilles, 2017; Okada et al, 2012).
A particular mention deserve the so-called PIRs (protein interaction reporters) that are trifunctional molecules containing the typical two domains with protein-specific reactivity plus a third additional functional group (usually biotin), advantageous for the enrichment of cross-linked products in the in vivo XL-MS experiments (Tang and Bruce, 2010).
In addition to the variety of chemical properties, the cross-linkers can be classified according also to another characteristic: the spatial organization of their atoms. Indeed, they can differ in the spacer arm lengths, which range from zero to tens of angstroms. The different lengths, acting as a molecular ruler, can be utilized to establish structural distance between very close or more distant residues.
Definitely, the combination of cross-linkers with different chemical and spatial features can provide more accurate data regarding the composition and dynamic of multiprotein complexes and PPI networks (Brodie et al, 2017; Ding et al, 2017).
XL-MS analysis: acquisition methods and bioinformatic tools
Each MS-based omic investigation, according to the features of mass resolution and mass accuracy available for the instrumental apparatus adopted, needs to establish a data acquisition method that may ensure the best performance for protein, peptide, or metabolite identification (Bilbao et al, 2015; Caterino et al, 2021; Costanzo et al, 2022c; Costanzo et al, 2020b; Defossez et al, 2021). Generally, MS-based instrumentations may perform their analyses in data-dependent acquisition (DDA) or data-independent acquisition (DIA) modes, or switching from one to the other in the newest high-resolution and accurate mass spectrometers (Costanzo et al, 2020a; Gallien and Domon, 2015; Lesur and Domon, 2015).
While the DDA strategy only selects the most intense precursor ions for fragmentation, DIA analysis allows the fragmentation of all precursor ions within a certain range of m/z and retention time (Jiang et al, 2022; Xin et al, 2022). Thus, it becomes intuitive that, when compared with DDA, the DIA approach allows an increase in the identification rate, even for low-abundant species, by producing a complete record of all the peptide types present in a sample.
It is known that DDA presents poor reproducibility for low-abundance proteins or peptides, thus not resulting ideal owing to the scarce bounty of cross-linked peptides (Gillet et al, 2012). Consequently, it lacks the throughput necessary to identify and quantify large fractions of entire cross-linked proteome, fundamental for full-scale XL-MS proteomics. However, even in the case of XL-MS studies, both DDA and DIA methods may be possibly applicable. Indeed, most of the XL-MS data present in the literature were obtained via DDA. To date, DDA is the method of choice for the structural studies of low-complex protein mixtures by requiring minimal dedicated assay development. In fact, cross-linked peptides detected by a conventional and robust DDA identification workflow provide useful information compatible for the interrogation of several downstream XL-MS dedicated software.
The challenge of determining in deep detail the landscape of in vivo PPI has as consequence the growing adoption of DIA as preferred method. With its high reproducibility in terms of identification and quantification, DIA represents the land of promise. Despite the potentiality of achieving detailed structural data, at present, DIA XL-MS is not hugely exploited, because of current software restrictions regarding the elaboration of cross-linking results. In their noticeable studies, Müller et al (2019) and Müller and Rappsilber (2020) tried to assess the issue, proposing an XL-MS workflow in which cross-linking data are acquired in DIA modality and, then, accommodated into Spectronaut, the leading software for DIA analysis (Martinez-Val et al, 2021). To the best of our knowledge, these represent the first attempts of applying the DIA method to an XL-MS approach.
It is mandatory to consider that the complexity of the MS/MS spectra of cross-linked peptides and the large number of theoretical reactions pose great challenges for the software elaboration. Standard shotgun proteomics tools are not optimal for cross-links identification because their MS fragmentation pattern differs a lot from the corresponding not cross-linked peptides, impacting tremendously on the possibility to control the false discovery rate (FDR). Thus, given the peculiarity of the XL-MS data, several dedicated algorithms have been introduced. The first generation of those programs makes use of approaches based on an in silico linearization of the cross-peptide pairs.
In detail, the strategy consists in the introduction into the search database of the chimeric linear protein sequences composed of all the theoretically possible combinations of cross-linked amino acids. The curated cross-link peptides database is then inspected with traditional search algorithms. Informatic tools such as xComb, Pro-Crosslink, VirtualMSLab, and other in-house implementations found their functioning on this concept (Gao et al, 2006; Koning et al, 2006; Leitner et al, 2010; Panchaud et al, 2010). Among these, SIM-XL was the first search engine capable of generating an interaction two-dimensional map of PPIs instead of the classical text files as results (Lima et al, 2018). As easily guessed, the direct consequence of their employment is the combinatorial explosion of the search space, due to all the peptide:peptide combos in the database being contemplated. Consequently, those cross-link analysis programs are limited to the search of a sequence database size of only a few proteins.
A partial solution to the increased search database size issue is offered by the xQuest algorithm. It is greatly popular for analyses in which the proteins are labeled with light and heavy isotope cross-linkers. Its operation relies on the detection of the isotopic couple at MS1 level, followed by the combination of sorted ions in a common cross-linked bond, whose prediction is accompanied by a scoring parameter (Rinner et al, 2008).
However, the majority of limits concerning the data elaboration step have been exceeded with the introduction of a set of programs, some of which are open-source and freely available. Indeed, this next-generation cross-linking-specific software tools appear to be amenable to many XL-MS experimental designs. Such algorithms, including Plink (Chang et al, 2015), StavroX (Götze et al, 2012), Xolik (Dai et al, 2019), MeroX (Iacobucci et al, 2018), OpenPepXL (Netz et al, 2020), and Kojak (Hoopmann et al, 2015), have been specifically designed for analyzing highly complex cross-linking data sets.
Due to their capability to greatly reduce the computational costs and times, once combined to the use of MS-cleavable reagents they accomplish the ideal workflow to apply in XL-MS proteome-wide investigations. Very recently, the novel application MaxLynx (Yılmaz et al, 2022) was integrated into MaxQuant, resulting efficient in the use of MS-cleavable as well as non-cleavable cross-linkers. In the MaxQuant environment, the Andromeda search engine was successfully tested for XL-peptides identification, whereas a novel dipeptide Andromeda score was optimized for efficiently computing the n-square problem for non-cleavable peptides. On the other hand, one of the general issues related to XL-MS workflows remains how to better assess the results, such as the visualization of the interactome network. To this purpose, a Cytoscape plugin (XlinkCyNET) has been recently released to handle XL-MS data for exploring large-scale protein interaction networks (Lima et al, 2021).
Quantitative XL-MS
XL-MS methodology is perfectly in line with the principles of quantitative biology, allowing the comparison of protein conformational variations between two or more conditions. In fact, QXL-MS is carried out by quantifying the cross-linked products to obtain a precise snapshot of the structural dynamics, enabling the assessment of interactome changes. Currently, compared with standard quantitative proteomics, QXL-MS makes use of diverse strategies to perform the analysis, such as the application of stable isotope labeling or label-free quantification (LFQ) approaches (Tang et al, 2021), showing that QXL-MS is as reproducible as other proteomic techniques (Chavez et al, 2015; Müller et al, 2018; Ross et al, 2004; Yu et al, 2016). In particular, the quantification step relies on the signals obtained from isotope-labeled cross-linkers, metabolic or isobaric labeling, or using peptide intensities in the label-free mode.
QXL-MS based on isotope-labeled cross-linkers is carried out by mixing the “light” and the “heavy” form of the same cross-linkers, which show differences in several atoms being usually replaced with their respective stable heavy isotopes. Samples to be compared are first labeled with one or the other cross-linker species and then combined generally at 1:1 ratio. After digestion and MS analysis, protein quantification is obtained according to the MS signal intensities produced (Schmidt et al, 2013).
Even in the case of metabolic labeling of cells, isotope-labeled peptides are employed for carrying on stable isotope labeling using amino acids in cell culture (SILAC)-based experiments before XL. In this approach, considering lysine residues as targets of both SILAC labeling and cross-linking reaction, isotope-labeled cross-linked peptides are formed as a consequence. As usual for SILAC, comparative analysis is performed by mixing at 1:1 ratio the cell protein samples from two different conditions grown in the light or heavy culture medium, and determining the differential abundance of proteins using the MS-based intensity of their signals (Munday et al, 2012). Even in combination with XL-MS, the MS intensities of the cross-links formed can be quantified as a measure of their abundance. In their brilliant work, Chavez et al (2015) performed an “edgotype” analysis by combining SILAC proteomics with in vivo XL-MS, detecting numerous quantitative interactome variations and protein structures in a cell model of chemoresistance.
Isobaric labeling, such as the tandem mass tags (TMT) and the isobaric tags for relative and absolute quantitation (iTRAQ) labeling, can support QXL-MS applications as well (Thompson et al, 2003; Yu et al, 2016). These strategies perform quantification at the peptide level; therefore, the sample preparation is independently performed for each sample until their mixing for LC-MS/MS analysis. In fact, unlike labeled cross-linkers and SILAC labeling-based QXL-MS that are usually circumscribed to binary comparisons (owing to scarce discrimination of isotope labels, spectra complexity, and reduced capability to detect low abundant XL peptides), isobaric labeling consents sample multiplexing and subsequent relative quantitation at the MS2 level (Yu and Huang, 2018).
This certainly favors the throughput and the reliability of quantification and reduces the sample complexity. However, with the TMT- and iTRAQ-based quantification being performed at the MS2 level, from the fragmentation of isobaric labels that generate unique reporter ions, quantitative inaccuracies may arise because all peptides are tagged with the same isobaric labels. A well-recognized caveat of isobaric multiplexing is the quantification interference due to co-isolation and co-fragmentation of ions that do not stem from the desired selected precursor ion (Gygi et al, 2019). Since ratio distortion caused by protein quantification interference is a common effect, MS3 (or MS n ) allows to minimize this interference (McAlister et al, 2014; Ting et al, 2011; Wühr et al, 2012).
Finally, the most likely preferred approaches for QXL-MS are based on LFQ, for lower costs and simpler preparation due to the absence of tags or labels to introduce. This approach may be utilized in combination with other structural approaches (Zheng et al, 2018) and is supported by informatic tools that increase the sensitivity of identification of XL peptides (Netz et al, 2020). In fact, the LFQ outcomes are often counteracted by the difficulties in the detection of cross-linked peptide signals, for the low abundance of cross-links in the samples. Nevertheless, the reproducibility of LFQ-based XL-MS adheres with the standards of reproducibility of the conventional quantitative proteomics (Müller et al, 2018).
LFQ analysis may be performed using different methods that rely on peptides intensity or spectral counts (Costanzo et al, 2018). LFQ XL-MS also holds the possibility to analyze samples using both DDA and DIA acquisition methods. Since DDA confers low reproducibility due to the low abundance of cross-linked peptides, targeted strategies, that is, multiple reaction monitoring (MRM) or parallel reaction monitoring (PRM), may be employed. For their nature, targeted techniques allow the rapid, sensitive, and reproducible detection of predefined sets of target peptides. The ability of triple quadrupole (for MRM) and quadrupole-orbitrap (for PRM) mass spectrometers to monitor several fragment ions of a selected precursor also provides accurate quantification (Costanzo et al, 2022b; van Bentum and Selbach, 2021).
In the context of XL-MS, MRM and PRM may serve to increase specificity and sensitivity for the quantification of less abundant cross-links (Chavez et al, 2016a; Peterson et al, 2012) and may be used as validation methods to confirm XL-MS results (Yu and Huang, 2018). Very recently, a PRM-based targeted approach combined with the use of the MS-cleavable DSSO cross-linker permitted the robust LFQ of selected cross-links and allowed to analyze the phosphorylation-dependent conformational dynamics of the human 26S proteasome (Yu et al, 2022).
Notwithstanding, the LFQ analysis performed in the DIA mode ensures more accurate results because of a better reproducibility of DIA when compared with DDA. These challenging perspectives of QXL-MS are in a constant way of development, with new QXL-MS workflows that are being developed and improved to include the DIA analysis in the routine of XL-MS. One study, in fact, reported a DIA-QXL-MS workflow that employs the Spectronaut software for the quantification of cross-links (Müller et al, 2019). Accordingly, the general problem that restricts the use and makes the analysis of QXL-MS data cumbersome is the availability of software and bioinformatic tools able to precisely analyze and extract the quantitative information in an automated manner. For this reason, the successful employment of QXL-MS in all system-wide projects still represents a challenging limitation (Piersimoni et al, 2022).
Structural Proteomics by XL-MS
Structural insights of protein complexes can be efficiently obtained by XL-MS. An overview of the XL-MS-assisted applications to the field of structural proteomics is schematically resumed in Figure 5. “Pure XL-MS” or alternatively “mixed-up XL-MS” approaches can support the characterization of protein structures and assemblies and the comprehensive definition of PPI (Serpa et al, 2012). In particular, the analysis of cross-links allows the identification of the protein residues that are involved in the formation of a given multi-subunit complex, providing precious information regarding the spatial placement and type of connections between the proteins.
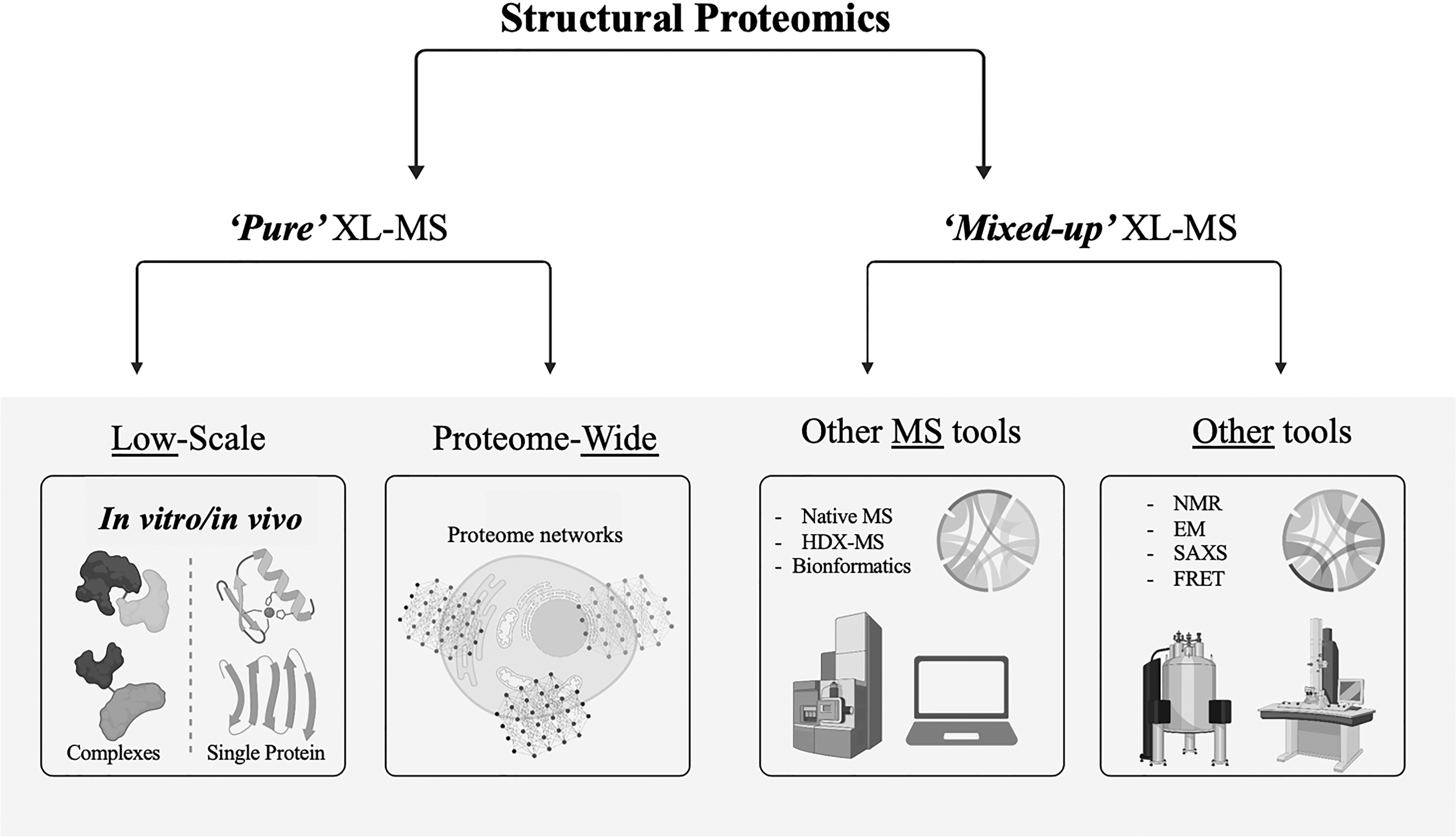
Structural proteomics embraces the application of XL-MS in low and large manner and its combination with other MS and non-MS workflows.
The knowledge of the points of contact of proteins within a multi-protein module are also important to define wider molecular interaction networks those modules are included into. Establishing a correct network topology better drives global network functions, not only describing the system but also predicting specific functions or phenotypes (Winterbach et al, 2013). Founding on such considerations, the architecture of a network easily takes its shape, paving the way to a variety of structural-based XL-MS experiments (Chen et al, 2010). In addition, the information retrieved from the protein cross-links becomes complementary to already existing structural studies, being able to corroborate existing data on structures, or providing new related computational models (Young et al, 2000; Yu and Huang, 2018).
Low-Scale XL-MS
The analysis of protein complexes by using XL-MS methodologies can be categorized in two subgroups, which include in vitro and in vivo experiments. The in vivo profiling of protein interactions is certainly the most appealing approach to resolve PPI and structures in their native cellular environments. Despite this, the majority of XL-MS studies have been narrowed to in vitro assays. The main reason behind this limitation is represented by the difficult detection of low-abundance cross-link products and their heterogeneity, thus forcing the systems to be scaled-down through the application of isolation and purification strategies to make the protein matrices simpler.
In vitro XL-MS analysis of protein complexes
Since the beginning of its application to drive biological problems, the study of protein complexes by XL-MS has been performed in vitro to define the molecular aspects of complexes architecture, but also their function or their regulative processes (Barysz and Malmström, 2018; Greber et al, 2015; Hurt and Beck, 2015; Khan et al, 2022; Sinz et al, 2015; Wang et al, 2017a). Usually, for such kinds of experiments, highly pure and homogeneous samples (e.g., recombinant proteins or protein complexes) have been largely adopted. From this point, a step forward was achieved by employing more heterogeneous samples, such as protein complexes purified by affinity from native cells.
In fact, according to this last strategy, many different affinity tags (e.g., His tag, biotin tag) have been developed over the years coupling efficient protein purification to XL-MS analysis. These tag-based affinity strategies were demonstrated to provide high selectivity and low background; this is particularly indicated for the biotin–streptavidin system, showing that streptavidin beads do not represent interfering reagents for the subsequent XL-MS analysis, suggesting improved power for the coupling of on-bead sample preparation and XL-MS to define protein complexes (Wang et al, 2017a; Wang et al, 2017b). By contrast, the application of antibodies to purify protein complexes is not favored in the case of XL-MS experiments because of their being sensitive to buffer conditions and their interference in peptide analysis (Yu and Huang, 2018).
In vivo XL-MS analysis of protein complexes
The technical improvements of XL-MS systems have paved the way to the large use of this technique for the in vitro analysis on single proteins or purified complexes, enabling their structural characterization. However, analyzing the in vivo connections and the topological features of multiprotein assemblies provides a drastically more significant biological value for interactomics studies. The biological relevance of in vivo cross-linking is reflected by the need to build precise PPI maps in a given time to dissect the specific molecular functions of interacting partners, in the context of the living cellular environment.
In vivo XL-MS holds the power to capture the dynamicity of an interactome that easily changes its topology and function in a cell (e.g., by PTMs, the changing environment), by “freezing” the existing protein connections through the cross-links. In fact, while robust and stable interactions can be more easily captured by commonly used MS approaches owing to their capability of surviving to diverse purification conditions, identifying the weak or transient interactions, and elucidating the function of their dynamic interplay within the cellular environment currently represent the most challenging aspect of interactomics science (Chavez et al, 2016b; Seath et al, 2021).
Notwithstanding, in vivo XL-MS is able to capture and characterize the structural dynamics of proteins in their native state, at the level of their surface regions accessible as recognition sites for the link with an interactor, which are mostly susceptible to the near environment variations. What is more, these accessible sites on the protein surface are more prone to become sites for PTMs, which in part may need to regulate the protein functions or their own interactions. As an example, protein phosphorylation and acetylation play a strong role on the modification of such sites, affecting with different implications the topology of the interactions (Bruce, 2012).
Following the dynamicity of the living proteome with all its shades represents a critical advancement for understanding the functions and the modifications of proteins and their networks in both health and disease (Altelaar et al, 2013; Costanzo et al, 2022a; Lundberg and Borner, 2019).
Proteome-wide XL-MS
Being able to reconstruct the spatial conformation and solve the structures of isolated protein complexes is certainly pivotal for the recognition of their function within their biological context. However, understanding the interplay between several protein assemblies and all the regulation mechanisms behind their assembly at a systematic level would represent a milestone of inestimable value. In fact, several technical improvements and developments of XL-MS systems have been achieved through the past decade, before their application as a tool for the proteome-wide discovery and identification of PPI networks. In fact, the increasing number of commercial cross-linkers and the availability of software and bioinformatic tools for data analysis have contributed to carry XL-MS from the analysis of simple protein samples to the deep and systematic characterization of PPI at the proteome level (Yu and Huang, 2018).
Beside the structural insights that are precious for the characterization of macromolecular systems, the XL-MS approach paves the way to a wider comprehension of the interaction profiles of whole proteomes, allowing to depict the in vivo protein network landscape (Wheat et al, 2021). A conceptual description would define as in vivo proteome-wide XL-MS those XL-MS studies in which the cross-linking reaction is performed before cell lysis. However, a more opportune term would be in situ XL-MS (Piersimoni et al, 2022).
With the advantage of needing small quantities of sample, XL-MS studies have largely spanned from the analysis of purified proteins and protein complexes to the exploration of organelles, living cells, tissues, and whole organisms (Liu et al, 2018; Navare et al, 2015; Ryl et al, 2020; Wheat et al, 2021; Wittig et al, 2021; Zheng et al, 2011). Despite the promising success, one big challenge that slows the application of XL-MS at the system-wide level is the huge complexity of samples and the dynamic range of the proteomes, in combination with other methodological limitations, such as the lack of optimized bioinformatic tools. To date, the maximum number of cross-links detected in proteome-wide investigations is around 10,000 (Piersimoni and Sinz, 2020).
One major bias of XL-MS is directed toward the high abundant proteins. In particular, this bias may be mostly triggered by the tendency of cross-links to get favorably established on high abundant proteins, rather than the lack in MS systems able to identify the cross-links on low abundant proteins. Besides, the optimization of certain parameters and protocols has resulted successful in increasing the detection of low-abundance proteins at proteome-wide level (Fürsch et al, 2020).
To deal with the enormous complexity of the proteome, the throughput of XL-MS-based systems has grown over the time. While permitting increased identification rates, on the other side, this would cause the generation of numerous false positive cross-links and the subsequent assignment of incorrect interactions. Previous research revealed that false positive cross-links are much more likely to be interprotein than intraprotein (Keller et al, 2019).
Hence, rigorous quality estimation and data validation are critical steps, whereas the conventional FDR may be prone to error propagation for cross-linking studies (Fischer and Rappsilber, 2017). In particular, the majority of the cross-linking investigations at the proteome level used to validate the XL-MS data of the discovered complexes with the data available for their corresponding tridimensional structures (O'Reilly and Rappsilber, 2018). It has been recently demonstrated that validating the cross-links using available 3D structures of representative protein complexes can drastically underestimate the error rate in proteome-wide XL-MS studies (Yugandhar et al, 2020).
Diminishing the complexity of the samples after cross-linking would provide the possibility to lowering the false positives in the case of proteome-wide studies, increasing the speed and the accuracy of identification of X-linked peptides, which are generally in lower amount compared with non-X-linked peptides. Certainly, the use of MS-cleavable rather than non-cleavable cross-linkers has increased the accuracy of the identification of X-linked peptides, taking advantage from the knowledge and recognition of their fragmentation patterns. Furthermore, reducing sample complexity is obtainable by performing enrichment steps at the protein and/or at the peptide level (Piersimoni et al, 2022).
Integrative Structural Proteomics
Combining XL-MS with other MS-based structural tools
Understanding the architecture of multi-protein units that associate within the cell permits to finely decipher the dynamics of a living system by using its proteome as analyzed from different sides (Costanzo et al, 2021b; Costanzo et al, 2020c). Proteome data will provide knowledge of the structures of these multi-protein complexes. The application of XL-MS runs the main advantage of inspecting the conformational topology of protein complexes, but still lacking on several details regarding the precise architecture of structures. These limitations are generated when the cross-linking alone, for its nature, is not able to derivatize a particular protein, owing to the specificity of the reagents for some protein residues and/or the corresponding accessibility of such residues in the protein. Furthermore, not always is prospected the possibility to decipher the sparse cross-linking data and to assign them to definitive structures, often delivering multiple possible conformations for a protein complex (Yu and Huang, 2018).
In such scenario, particularly winning may be the choice of combining XL-MS with other MS-based structural tools. This process is to be carried out by collecting several high-quality MS data, translating such information into spatial restraints and, then, employing the restraints to recreate a group of structures, aided by bioinformatics and computational modeling tools (Alber et al, 2007; Leitner, 2016; Sali et al, 2003; Slavin and Kalisman, 2018). In example, the combination of a bottom-up approach for X-linked proteins and the top-down analysis by native MS has resulted in the successful determination of the tridimensional structure of various multi-protein assemblies (Glas et al, 2015).
This complementary approach is based on the capability of native MS to define the global complex topology of complexes and the subunit stoichiometry, and on the attitude of XL-MS to precisely recognize the interaction sites between the proteins with a conservative effect on the dynamic interactions. This kind of combined approach is still limited by the fact that endogenous complex assemblies are difficult to be extracted from a sample in an adequate amount and purity conditions to be analyzed by native MS (Yu and Huang, 2018).
An additional MS-based method for obtaining structural insights on molecules is the hydrogen–deuterium exchange mass spectrometry (HDX-MS). HDX-MS is able to analyze the degree of disorder in proteins by monitoring the rate of in-solution exchange of hydrogen atoms with deuterium at the amide backbone of proteins. Such approach allows to capture the dynamics of a protein or protein complex detecting the conformational changes at single amino acid resolution, thus providing information on protein secondary and tertiary structures, and on allosteric regulative mechanisms (Kochert et al, 2018; Konermann et al, 2011; Pacholarz et al, 2012; Piersimoni et al, 2022).
HDX-MS has been found to finely complement with XL-MS, owing to the ability of the latter to also capture low-affinity or transient interactive connections between proteins. One brilliant example of such integrative application has been recently published as for the current COVID-19 pandemic research. In particular, the dynamics of SARS-CoV-2 full-length NSP7 and NSP8 proteins and the NSP7:NSP8 protein complex have been elucidated. These findings validated the sites of interaction as already published for the 3D heterotetrameric crystal structure (Courouble et al, 2021). In addition, it was found that NSP7 and NSP8 do not assemble into a hexadecameric structure as implied by the SARS-CoV-2 full-length NSP7:NSP8 crystal structure (Courouble et al, 2021). Biologically meaningful was evaluated the inhibition of the interaction of NSPs proteins as one of the strategies to control SARS-CoV-2 infection, besides mass vaccination (Costanzo et al, 2021a; Sarma et al, 2022).
Combining XL-MS with other structural tools
Researchers have expanded the frontiers of cellular and molecular imaging by developing several analytic tools and software to obtain structural information in a plethora of biological contexts (Bai et al, 2015; Costanzo and Costanzo, 2022). In particular, various techniques have been used so far for finding structural details on proteins or protein complexes, including X-ray crystallography, NMR spectroscopy, cryo-EM, small-angle X-ray scattering, and Förster resonance energy transfer. The main advantage of such approaches is the possibility to capture high-resolution 3D structural details on proteins and, possibly, on their connections in space (Bartesaghi and Subramaniam, 2009; Ziegler et al, 2021).
In particular, cryo-EM has come out as an elective method in structural biology, providing structure details of protein assemblies at the near-atomic resolution, especially for its less laborious sample preparation if compared with X-ray crystallography and NMR. While the latter two produce high-resolution results, the advantages of cryo-EM slip away owing to the low resolution in EM maps produced by those areas with unassigned electron densities (Earl et al, 2017; Yu and Huang, 2018). As a consequence of its low resolution, especially when complex protein assemblies are analyzed, cryo-EM may generate uncertain models and subsequent misinterpretation of 3D structures. In this scenario, orthogonal information to the low-resolution EM maps may be offered by complementation with XL-MS data at the amino acid level (Greber et al, 2014).
If different conformations of a heteromeric complex are equivalent according to the produced cryo-EM map density, more than one model for that structure is proposed. Actually, the precise length of the cross-linkers and their covalent bond to the functional groups of the side chains of amino acids permit their identification by MS, dictating distance constraints on the 3D structure of proteins or protein assemblies. In fact, XL-MS allows accepting or rejecting one of the models proposed by the cryo-EM density maps, with the identification of unique and specific cross-links between the interacting residues of two protein subunits (Piersimoni et al, 2022).
Intrinsically disordered proteins
As widely acknowledged, globular, transmembrane, and fibrillar proteins have unique tridimensional structures, which assist their functions in physiological conditions. Apart from these examples, there exist other proteins not characterized by unique or stable 3D structures, whose conformations are flexible in space and highly dynamic even under physiological conditions (Turoverov et al, 2010; Uversky and Dunker, 2010; Wright and Dyson, 1999). Such proteins or protein regions, defined as intrinsically disordered proteins (IDPs) and intrinsically disordered regions (IDRs), respectively, represent a consistent portion of the eukaryotic proteome, which nevertheless accomplish several, even important cellular functions.
According to bioinformatic predictions, up to 30% of the proteome contains intrinsically disordered structures, with nonstable secondary or tertiary conformations (Peng et al, 2015; Piersimoni et al, 2022). Their structural flexibility may provide advantages more than limitations to the adaptability for the binding with other molecules or their functions (Fuxreiter, 2019; Oldfield and Dunker, 2014; Wright and Dyson, 2015).
One challenge in structural biology is to assign correct structures to those IDPs with no defined or univoque structures defining the unfoldome. Even because IDPs and IDRs are frequently associated with pathological conditions (Dunker et al, 2008; Uversky et al, 2009), the interest in their folding mechanisms and involvement in disease has constantly grown, leading to their consideration as possible therapeutic targets.
Thus, structural MS is being considered a key instrument for the structural and functional characterization of IDPs and IDRs in the unfoldomics era (Beveridge and Calabrese, 2021; Beveridge et al, 2014; Mitra, 2021). Generally, XL-MS provides information regarding the conformational status of assemblies analyzing the cross-links formed during the chemical reaction. Therefore, the details corresponding to the distance constraints extracted from XL-MS data are analyzed for the modeling of IDPs and IDRs (Piersimoni et al, 2022). For such scopes, ion mobility (IM)-MS has been used in combination with XL-MS, providing increased sensitivity for the identification of cross-links (Ihling et al, 2021; Schnirch et al, 2020), suggesting that many other MS tools may be combined to obtain meaningful results. Accordingly, findings on the protein conformers in solution, on the transitions from different conformations, and the disorder degree may be obtained using both native MS and IM-MS (Beveridge et al, 2014; Kaltashov and Mohimen, 2005; Stuchfield et al, 2018).
Native MS demonstrated that the normal distribution of the states of charge, which define the protein solvent-accessible surface area, correlates with protein structure. In fact, it was shown that proteins with a stable folding have more compact distributions of their states of charge, when compared with disordered ones (Natalello et al, 2017; Santambrogio et al, 2019). The use of IM-MS has also demonstrated a similar direct correlation between the rotationally averaged collision cross sections of proteins in their ionized form and the degree of compactness of proteins and protein assemblies conformations, even in the absence of solvent (Beveridge et al, 2014; Stuchfield et al, 2018). Thus, IM-MS-based applications acquire importance in exploring the conformational status of proteins, determining whether they are present as structured or disordered.
Conclusions and Perspectives
XL-MS has refashioned the principles of functional and structural biology with the delivering of new knowledge in the dynamics of proteome interactions and protein complexes architectures, as for mapping PPI in the context of global functional networks. Therefore, investigating protein interactions by XL-MS is contributory in discovering also the finest functional connections for both healthy and disease research. In fact, with its initial application to purified proteins and protein complexes, the XL-MS methodology has greatly developed allowing the analysis of the systematic interactions even at the proteome-wide level. In addition, one of the promises of XL-MS applications is to support, as complementary and integrative approach, the already existing methodologies developed for elucidating structural insights such as the cryo-EM, X-ray crystallography, NMR, or other MS-based techniques such as native MS and HDX.
On the flip side, some research is still needed to identify precise workflows, which may combine the use of suitable cross-linkers and efficient computational tools for the downstream data analysis. In example, the use of non-cleavable cross-linkers is preferred for the study of purified proteins or simple protein assemblies, whereas MS-cleavable cross-linkers result more advantageous for proteome-wide XL-MS investigations. Based on these concerns, harmonizing the procedures and the protocols, from sample preparation and data acquisition to the data elaboration, is essential to guarantee the generation of reproducible results and reliable findings to be shared among researchers. To this aim, the first community-wide XL-MS study has made an effort to establish some guidelines and generally accepted procedures to drive the harmonization of XL-MS-based experiments (Iacobucci et al, 2019).
Intriguingly, the applicability of the XL-MS concept appears not limited solely to the protein interactomics field but presents the potentiality to reconstruct virtually all the possible cross-talks between proteins and other molecules, such as nucleic acids or metabolites. Currently, the XL-MS application is a palpable reality capable of describing high-resolution structural connections for RNA-proteins macromolecular complexes (Sarnowski et al, 2022). To profile protein–metabolite interactions, MS strategies (e.g., IM-MS, limited proteolysis-coupled MS) are also explored (Beveridge et al, 2016; Qin et al, 2020), but none of the proposed workflows seems to be applicable to intact cells, limiting their ability to assess in vivo relevance of these interactions. Thus, the determination of a chemoproteomic network in a system-wide manner might be an excellent test bed for the XL-MS methodology in the near future. With the actual technical issues mostly being borne, we expect a great rise in the number of functionally reconstructed XL-maps of all possible molecule combinations.
Footnotes
Authors' Contributions
Author Disclosure Statement
The authors declare that they have no conflicting financial interests.