
Abstract
Colorectal cancer (CRC) is reportedly the second leading cause of cancer death worldwide. By the end of the decade, there will likely be more than one million fatalities worldwide from this cancer, with an estimated 2.2 million additional cases. We need new ways of thinking about cancer research. One approach is to deploy systems science using quantitative proteomics to obtain postgenomic and functional insights into cancer. The present study compares the tissue proteome of CRC (n = 10) with the matched peritumoral controls (n = 10) in samples obtained from the Indian subcontinent. When compared with the controls, a list of 22 substantially altered protein candidates was identified, which were associated with the growth, survival, and metastasis of the tumor. A list of the unique peptides from top significant proteins, including olfactomedin-4, alanyl aminopeptidase, and grancalcin was further validated using a parallel reaction monitoring-based targeted proteomics approach. In addition, biological pathway analysis showed perturbation in key biological processes, including dysregulation in purine metabolism, MYC targets in cancer, DNA repair, and replication, and leukocyte transendothelial migration, among others. The protein panel reported herein is also shown to be dysregulated in CRC and warrants further research toward understanding pathobiology, diagnostics, and therapeutics development in CRC.
Introduction
Colorectal cancer (CRC) remains among the deadliest malignancy across the globe (Barpanda et al, 2022). It stands to be the leading cause of cancer mortality worldwide. It is the second most common cancer diagnosed in women and the third most in men and accounts for ∼10% of cancers diagnosed annually (Dekker et al, 2019). The global burden of the tumor is estimated to be around 2.2 million new cases with more than 1 million deaths by 2030 (Rawla et al, 2019). For example, in India, CRC cases are among the top five most frequent cases reported. According to the population-based cancer registry's time trend report, the disease has a markedly rising trend in all of the major cancer registries, with annual percentage changes ranging from 0.9% to 5.8% for the colon and 2.7% to 9.8% for the rectum (Behera and Patro, 2018). India has one of the lowest rates of CRC 5-year survival worldwide. Indeed, the global comparison of population-based cancer survival (CONCORD-2) study shows that the 5-year survival rate for CRC in India is decreasing (Patil et al, 2017).
We need new ways of thinking about research on CRC specifically, and cancer more generally around the world. One approach is to deploy systems science using quantitative proteomics to obtain postgenomic and functional insights into cancer. Moreover, the majority of the current methods for diagnosing CRC involve invasive procedures, including colonoscopies, biopsies, and other radiology-based methods (Vega et al, 2015). There is also a paucity of research on quantitative proteomics in populations outside North America and Europe, which are sorely needed to elucidate the molecular pathways underlying the neoplastic changes of the normal colon mucosa into malignancy. Oncologists in India, for example, have noted that, when compared with instances reported internationally, the majority of CRC patients in India appear to be at younger ages, with more advanced disease, signet ring shape, and anorectal rather than intestinal sites of origin (Patil et al, 2017). The present study reports a high-throughput quantitative tissue proteomics approach to study the molecular substrates of CRC, using a tandem mass tag (TMT) to identify potential biomarker candidates.
The altered proteins identified herein were also subjected to biological pathway analysis to decipher new insights into CRC disease biology. While these initial findings need further corroboration in independent studies, the study is one of the first comprehensive proteome-wide investigations of patients with CRC in India, and contributes to the global efforts to unpack CRC pathophysiology through a systems science lens. The data also inform future diagnostics and therapeutics discovery research.
Materials and Methods
Clinical tumor sample
Clinical tumor tissue samples and healthy peritumoral tissues were collected from KEM Hospital, Mumbai. Immediately following the surgery, fresh clinical tissue samples were brought to Indian Institute of Technology Bombay, and preserved at −80°C until use (Supplementary Table 1). For the quantitative proteomic analysis, the study employed clinical colorectal tumor samples and matched peritumoral noncancerous control samples from individuals 48 to 63 years of age. The study is approved by the Indian Institute of Technology, Bombay, Ethics Committee (IITB-IEC/2021/006) and a written informed consent was obtained from all study participants.
Protein extraction and sample preparation for quantitative proteomic analysis
Around 50 mg of tissue sample was weighed and rinsed with chilled 1 × phosphate-buffered saline twice to wash the blood contents. The tissue sample was lysed using 300 mL of lysis buffer containing 8 M urea, 50 mM tris pH 8.0, 75 mM NaCl, and 1 M MgCl2 and sonicated followed by bead beating, to make tissue lysate. Centrifuging the debris at 8000 g for 15 min at 4°C separated it from the lysate. The supernatant was collected in a new, sterile 1.5-mL low-binding tube (Biswas et al, 2021). Using standard amounts of bovine serum albumin, these tissue lysates were quantified following the Bicinchoninic Acid Assay method. For protein digestion, the protein lysate of each sample was reduced with tris (2-carboxyethyl)phosphine at 37°C for an hour. Additionally, the alkylation process was carried out using iodoacetamide at room temperature for 20 min in dark. The tissue lysates containing 8 M urea were further diluted five times in volume with a dilution buffer (25 mM Tris pH 8.0 and 1 mM CaCl2). Trypsinization was done by Pierce trypsin in a 1:30 ratio followed by incubation at 37°C in a dry bath for 16 h (Mukherjee et al, 2020). On-site C18 filter stage tips were used to further desalt the dried samples.
TMT labeling and high pH reversed-phase peptide fractionation
TMT six-plex isobaric tags (TMTsixplex™ Isobaric Label Reagent Set, 1 × 0.8 mg; Catalog No.: 9006; Thermo Fisher Scientific) were used for the labeling of the digested CRC peptides as per the standard protocol. In a nutshell, the peptides were reconstituted in a dissolution buffer and mixed thoroughly. Each patient sample was combined to create a pooled sample, which served as a standard for normalizing the mass spectrometric data. Dried TMT labels were reconstituted using anhydrous acetonitrile (ACN) followed by tagging of the digested sample. For effective labeling, 15 μg of each digested peptide sample was labeled followed by proper mixing and incubation for 1 h at room temperature (Kumar et al, 2020a). All of the corresponding samples were pooled and mixed thoroughly and dried in a vacuum centrifuge. Following the manufacturer's directions, samples were subsequently separated into four fractions using high-pH reverse-phase chromatography (Pierce™ High pH Reversed-Phase Peptide Fractionation Kit; Catalog No.: 84868; Thermo Fisher Scientific).
Liquid chromatography–mass spectrometry/mass spectrometry analysis
The multiplexed TMT-labeled CRC samples were analyzed using an Orbitrap Fusion Tribrid mass analyzer coupled with an Easy-nLC 1200 liquid chromatography system (Thermo Fisher Scientific). The mobile phase was made up of MS-grade water with solvents A and B that were 0.1% formic acid (FA) and 80% can, respectively. Each labeled fraction was reconstituted with 0.1% FA and 1 μg peptide was injected into the nano analytical column (Thermo Fisher Scientific). The peptides were eluted gradually with an isocratic gradient of 10–35% B in 103 min, 35–95% B in 2 min, and 95% B for 15 min at a constant 300 nL/min flow rate. Data-dependent MSn survey scans were carried out in the orbitrap mode with higher-energy collisional dissociation (HCD) following ion isolation with the quadrupole. The orbitrap MS method was set with a scan range of 375–1700 m/z at a resolution of 60,000. The automatic gain control (AGC) target was set at 5e4.
The MS/MS acquisition method was set as 20 MS/MS scan; 30,000 resolutions; AGC of 4e5; and an isolation width of 1.2 m/z. The normalized collision energy was optimized at 35% with a dynamic exclusion window of the 40s. A lock mass option from ambient air (m/z 445.1200025) was used for internal calibration (Verma et al, 2021).
Database search and MS stats analysis
The Raw files from quantitative liquid chromatography–mass spectrometry/mass spectrometry (LC-MS/MS) analysis were processed using Proteome Discoverer (PD) version 2.4 (Thermo Fisher Scientific). All fractions were combined and searched using SEQUEST HT and Mascot (v2.6.0) search engine against the UniProt human database (Proteome ID: UP000005640, Organism ID: 9606). The dynamic modifications were set for the TMT reagents (+229.163 Da) on lysine and N-termini along with the oxidation of methionine residues (+15.9949 Da). A Static modification was kept as carbamidomethyl (+57.021 Da) on cysteine, and monoisotopic masses, and a maximum of 2 missed cleavages were allowed for digestion. The mass tolerance for precursor and fragments were set as 10 ppm and 0.05 Da, respectively, and the false discovery rate (FDR) was kept at <1%. The MSstatsTMT package in R has been used to summarize the proteins at the protein level and analyze their significance (Huang et al, 2020). Using an in-house R-code, the peptide spectrum matches (PSMs) were preprocessed in PD and then transformed into the necessary input format for MSstatsTMT.
Using the “protein summarization” script, the protein abundance was determined based on the quantification of the peptides (MSstatsTMT). The peptide-level data were integrated into protein-level data after normalization between MS runs using reference pool channels and the imputation of missing values. The accelerated failure model was used by MSstats to impute missing values in the protein summarization method, where it was assumed that missing values were censored. The method was set as “MSstats” in the protein summarization method with both global and reference normalization set as “TRUE.” A moderated t-test was then performed on the normalized values using the “Group-Comparison-TMT” function. Proteins passing the threshold of p value <0.05 and logFC >1.2 were considered significant and used for further biological analysis.
Validation of significant proteins using parallel reaction monitoring-based targeted proteomics analysis
The significantly altered proteins were validated using parallel reaction monitoring (PRM)-based targeted proteomics using an Orbitrap Fusion mass analyzer (Thermo Fischer Scientific) at MASSFIITB, Indian Institute of Technology, Bombay. PRM method development and optimization of target proteins were performed using Skyline daily. The PRM assay was run with a method duration of 40 min containing a full scan followed by quantitative MS/MS mode separately. The full scan MS OT mode was kept at a resolution of 60,000, a scan range of 350–1700 (m/z), and a maximum injection time of 50 ms, and the tMS2 OT HCD having Isolation Window 1.2, resolution of 15,000, Scan Range of 50–2000, and maximum injection time of 40 ms (Kumar et al, 2020b). The isolation list of the selected proteins was generated from Skyline daily with the Human database (Version: 2020_04) and the PROSIT library was used for a spectral match. One microgram of the peptide was injected into nLC having a liner gradient of 40 min with solvent A comprising 0.1% FA in water and solvent B comprising 80% ACN in 0.1% FA water and a flow rate of 300 nL/min.
Isolation list preparation
The protein list's UniProt ID was used as an input for isolation list preparation using Skyline daily. The peptide sequences for these proteins were obtained from the peptide output file and examined in the neXtProt peptide uniqueness checker to ensure that the peptides were unique. The unique peptide sequences were imported into Skyline daily and an isolation list was created with peptides having 8–21 amino acids with 0 missed cleavage. The isolation list, including both precursors and product ions of selected peptides for Orbitrap Fusion was exported and imported to the PRM method as discussed above and analyzed further.
Molecular pathway analysis
The list of significant differentially expressed proteins (DEPs) in CRC in comparison to the matched control was taken forward as an input for all the biological and functional analyses. Gene Set Enrichment Analysis (GSEA; borad.mit.edu/gsea) was performed where the KEGG pathway database under canonical pathways was used from Molecular Signatures Database (MSigDB). The number of permutations was set to 1000 and the metric for ranking genes was set to Signal to Noise. Network enrichment analyses were performed in STRING, where “confidence” was chosen as a parameter of network edges and the interaction score was kept as default. The biological clustering was done using functional enrichment analysis. The entire procedure followed starting from sample preparation to pathway analysis is illustrated (Fig. 1A–F).
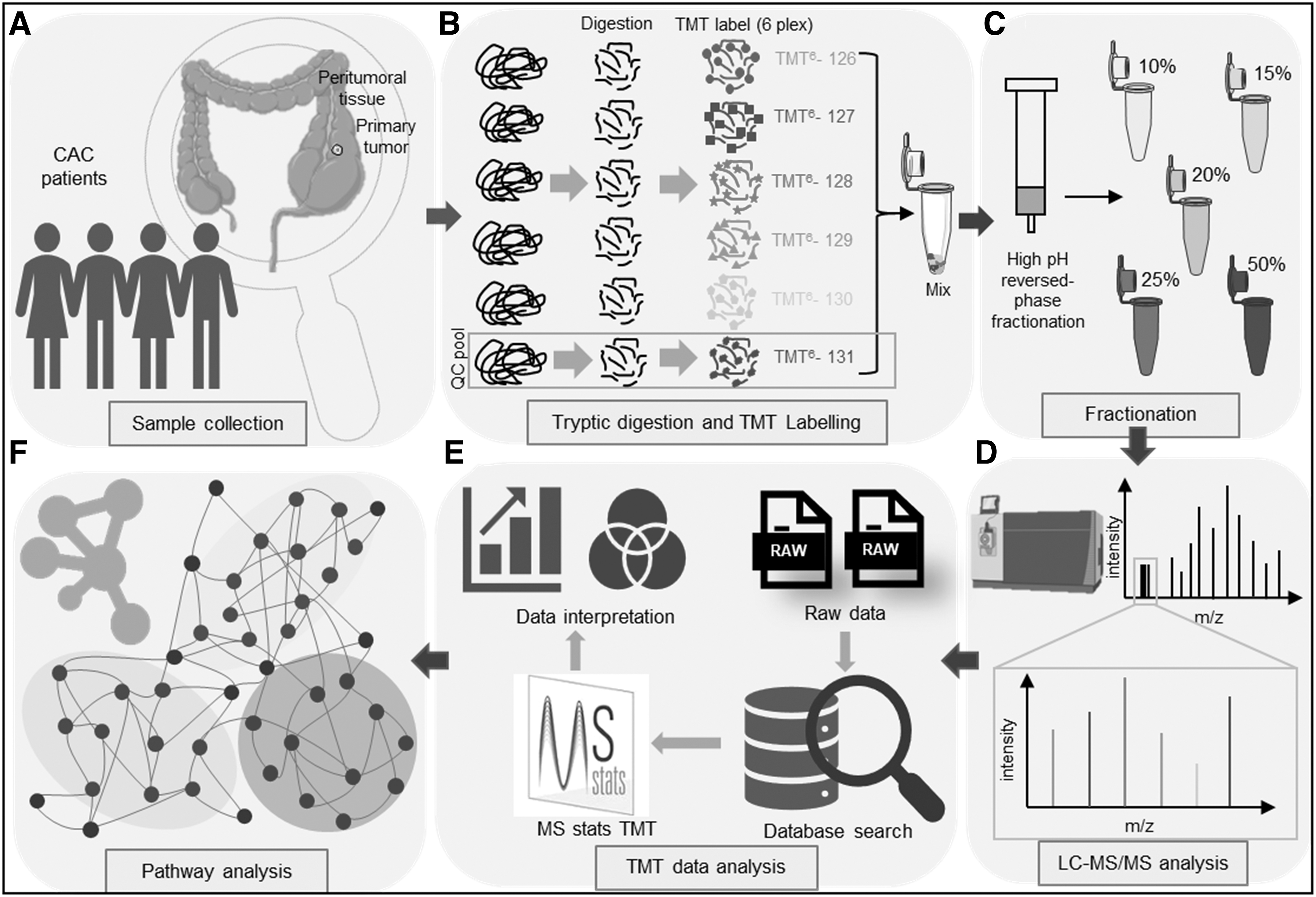
Schematic representation of the experimental strategy used for quantitative proteomic analysis and data analysis.
Data availability
The data are uploaded to the ProteomeXchange Consortium with ID-PXD039285. The codes used in the analysis can be accessed in the GitHub (https://github.com/AnkitHalder530/CAC_MSStatsTMT).
Results
Identification of DEPs in TMT-labeled colon adenocarcinoma tissue lysates
Trypsin digestion of the CRC tissue samples and their corresponding peritumoral control tissue lysates was followed by TMT labeling. Five of the individual sample lysates were labeled with the TMT labels 127, 128, 129, 130, and 131, and a pool of all the samples (n = 20) was labeled with the TMT label 126 in each of the six-plex reactions. The labeled peptides were pooled followed by a high pH reversed-phase fractionation to reduce the sample complexity. The fractionated samples were later subjected to LC-MS/MS-based analysis. The raw files obtained from the LC-MS/MS analysis were further analyzed independently using a PD. A total number of 2508 proteins were identified having at least one unique peptide. The identified proteins were filtered and proteins having greater than 1 unique peptide is taken for statistical analysis using the MS stats package for TMT-labeled data analysis. Furthermore, datasets were normalized and log-transformed and a pairwise t-test was performed (Fig. 2A; Supplementary Fig. S1).
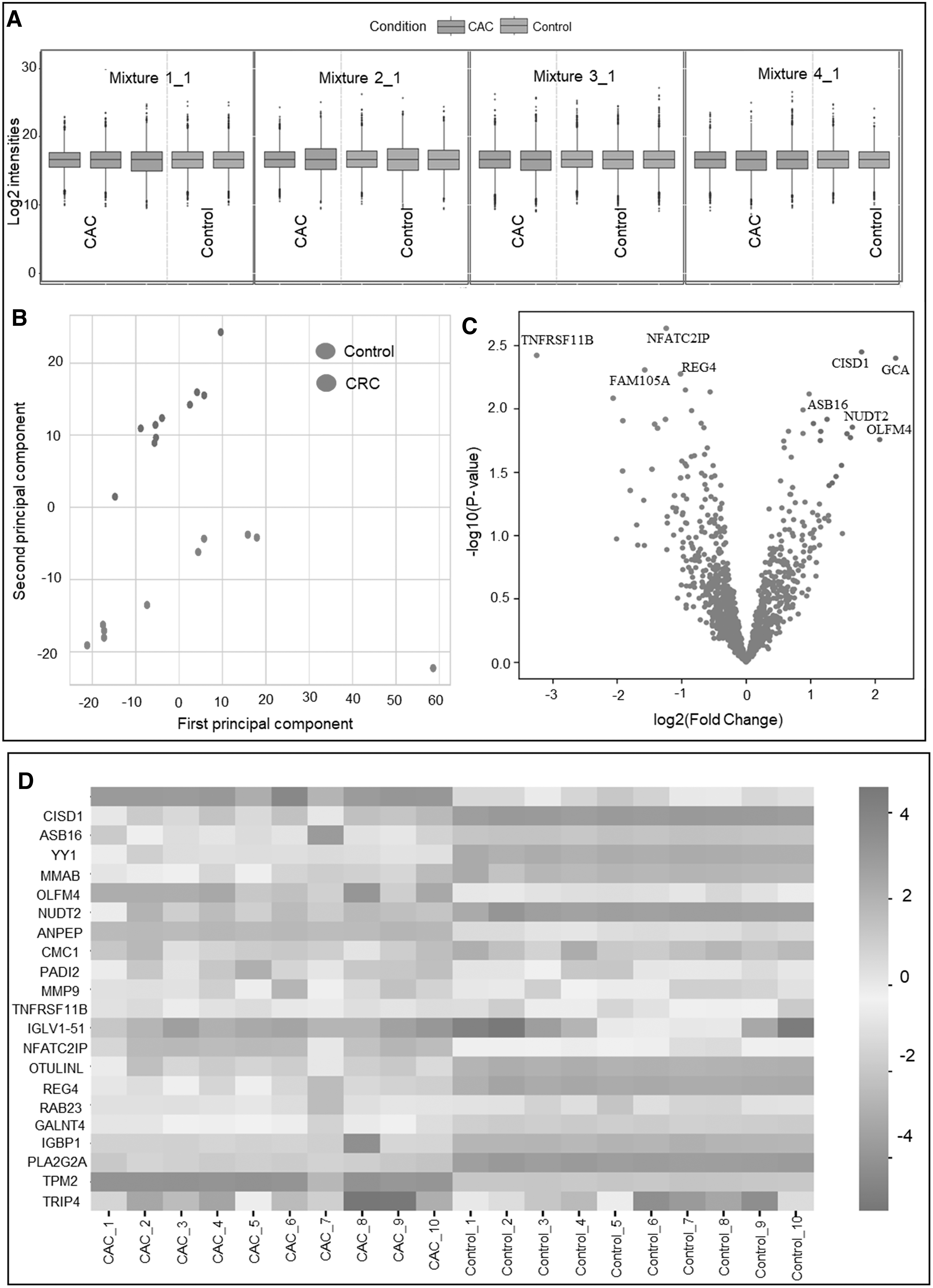
Comparative quantitative proteomic analysis of colorectal cancer and peritumoral tissues.
Again, the samples were analyzed using principal component analysis to assess the overall change between the CRC and control sample cohorts (Fig. 2B). Considering the significance criteria of a p value <0.05 along with log2FC of 1.2, we identified 22 dysregulated proteins. Out of these 11 proteins each was identified to be significantly upregulated and downregulated in the disease. The two sample cohorts were seen to have separated into different groups. Significantly varied protein expression between the two cohorts is shown in the volcano plot (Fig. 2C). A list of important proteins with differential expression was found by statistical comparison of the cohort, as represented by a Heatmap (Fig. 2D).
Proteins, including grancalcin (GRAN), CDGSHM iron–sulfur domain-containing protein-1, Ankyrin repeat and SOCS box protein 16, transcriptional repressor protein Yin Yang 1 (YY1), olfactomedin-4 (OLFM4), Bis(5′-nucleosyl)-tetraphosphatase (NUDT2), alanyl aminopeptidase (AMPN), protein arginine deiminase type-2 (PADI2), and matrix metalloproteinase-9 (MMP9) were seen to be significantly upregulated in CRC. In addition, proteins, including Activating signal co-integrator 1 (TRIP4), Phospholipase A2 (PLA2G2A), Polypeptide N-acetylgalactosaminyltransferase 4 (GALNT4), Regenerating islet-derived protein 4 (REG4), tumor necrosis factor (TNF) receptor superfamily member 11B (TNFRSF11B), etc. were identified to be downregulated (Supplementary Table 2). Comparative visualization of the dysregulated proteins is represented as rain cloud plots (Fig. 3; Supplementary Fig. S2).
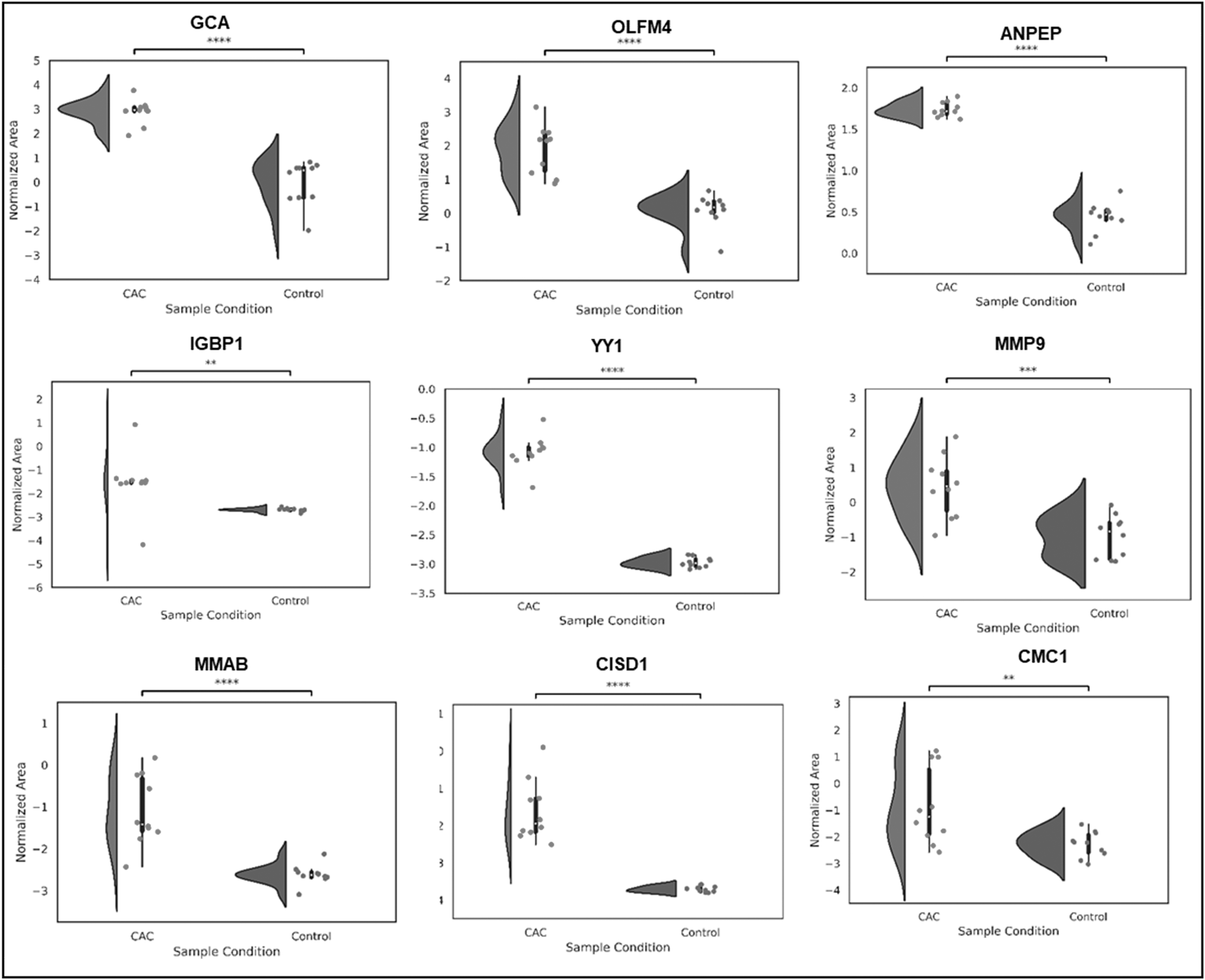
Comparative expression of significant proteins in CRC and controls. Figure showing the abundance of significantly upregulated protein panel, including GCA, OLFM4, ANPEP, IGBP1, YY1, MMP9, MIMAB, CISD1, and CMC1.
PRM-based validation of significant proteins
A PRM-based targeted proteomics study was performed to validate dysregulated proteins identified from deep quantitative proteomic analysis of CRC. One microgram peptide from each of the samples was injected into the analytical column and the significant proteins, including OLF4, AMPN, and GRAN, were monitored to gain more confidence in the discovery phase data (Fig. 4A–C). The peptide spectra were further matched with the PROSIT library and a score of >0.7 were considered for further analysis. As per the result of the quantitative proteomics approach, all these proteins were found to be of similar trend validating their relevance to the disease. The peak area of each peptide was considered for performing the t-test using the MS stats package in Skyline. Significant criteria included a p value of <0.05 and a Log2FC of 1.2 for validation of the dysregulated proteins. From the analysis, all the mentioned proteins along with their peptides were identified to be significantly dysregulated validating the trend of discovery of proteomics data.
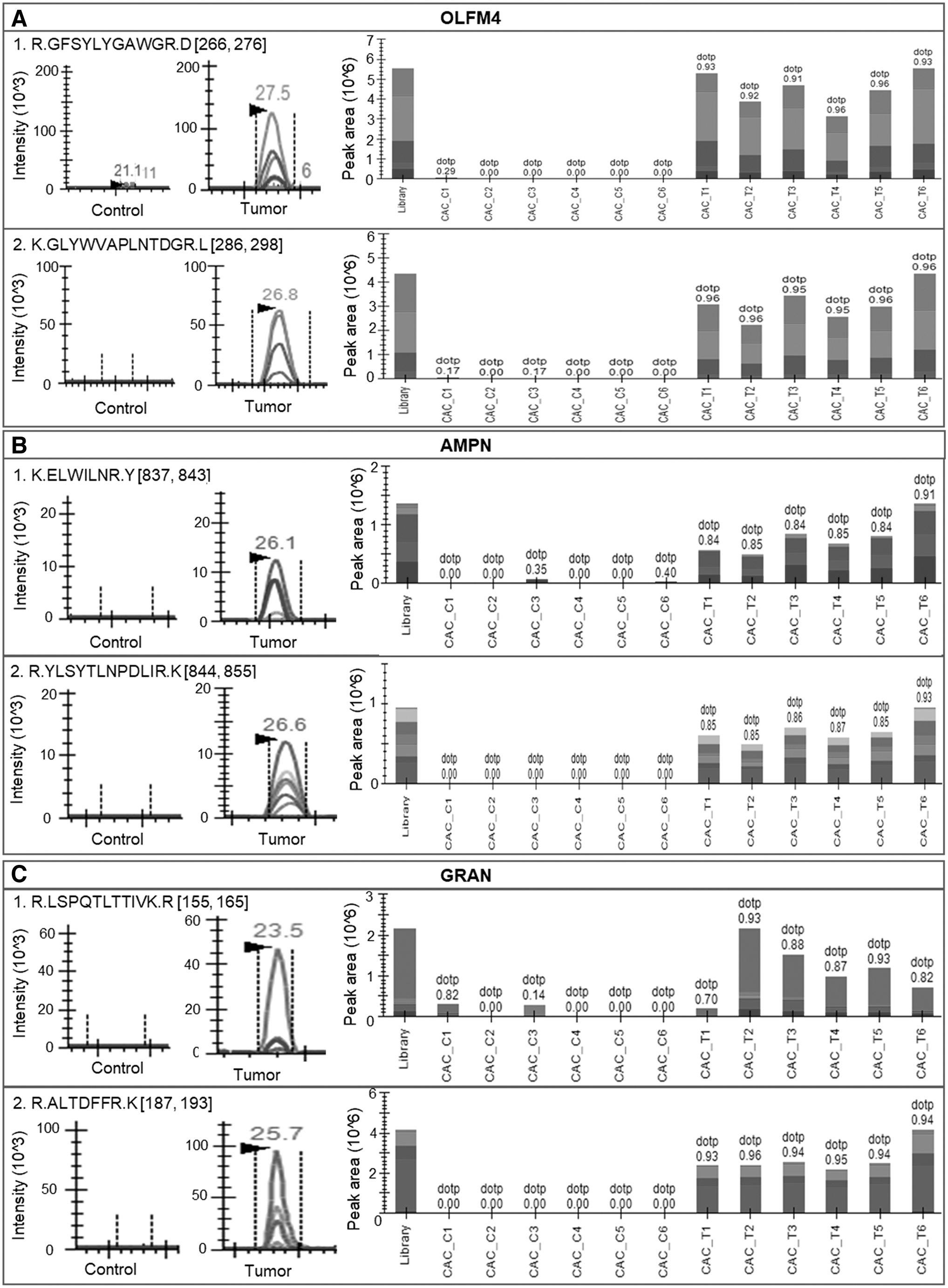
Parallel reaction monitoring-based validation of important proteins altered in CRC. Peptides of the top significant proteins were monitored using parallel reaction monitoring.
Biological pathway analysis and GSEA
The analyzed files were further processed, and the normalized intensities were used as input for GSEA. The analysis showed a significant dysregulation in the leukocyte transendothelial migration processes with the normalized enrichment score of 1.6 having a nominal p value of 0.0005. Purine metabolism was identified to be dysregulated with a normalized enrichment score of 1.4 and a nominal p value of 0.01 (Fig. 5A). Additionally, the significantly dysregulated pathways were analyzed and shown in the circos plot (Fig. 5B; Supplementary Fig. S3A, B). GSEA results were filtered with a minimum enrichment score cutoff of 1 and an FDR Q value of <1. The enrichment analysis revealed dynamic alteration in the cellular signaling pathways comprising leukocyte transendothelial migration, purine metabolism, apical junction, MYC target, DNA replication and repair, RNA processing localization, and regulation of protein localization.

Biological pathway analyses.
The leukocyte transendothelial migration was shown to be severely dysregulated in CRC, with considerable changes to key elements such as SIPA1, MMP9, RHOA, and F11R. Proteins linked to the hallmark MYC targets were identified to be positively enriched with proteins mapping to the pathway, including MCM5, CCT4, IMPDH2, XPO1, etc. A similar upregulated trend was also observed in the process of DNA repair where DDB1, POLR1C, and POLR1B were mapped. Pyruvate metabolism is identified to be significantly upregulated in the CRC group with positive enrichment of LDHA, ME2, and DLAT, MDH. Similarly, important proteins from Purine metabolism, including NUDT2, AK4, and GART, were found to be dysregulated in the disease.
Furthermore, the protein–protein interaction network revealed the dysregulation of major biological signaling pathways linked to cancer metabolism and homeostasis. The biological analysis and network visualization were done using Cytoscape V3.9.1. In the earlier GSEA analysis, similar pathways were also observed to be dysregulated. The apical junction along with leucocyte transendothelial migration was identified to be perturbed in the disease. DNA replication and repair, messenger RNA (mRNA) processing, and cell cycle process were also identified to be altered. A major hub connecting the ribose phosphate metabolic process was identified, where the majority of the interactors were identified to be positively expressed in CRC. Along with the purine metabolism, several other biological pathways, including localization of RNA and protein, long-chain fatty acid import, and CCL 18 signaling pathway were identified to be dysregulated. Additionally, gene and protein expression by the JAK-STAT signaling pathway along with the central carbon metabolism was seen to be upregulated in the disease (Fig. 5C).
Discussion
With a relatively high fatality rate, colon cancer continues to be among the most terrifying diseases in the world. Despite impressive developments in CRC research, the management of the condition is still a major concern. The current screening methods for CRC include colonoscopy, fecal occult blood testing, flexible sigmoidoscopy, double-contrast barium enema, virtual colonoscopy, and fecal DNA testing. However, these invasive tests only identify cancer when the tumor is advanced and reached a large size. Hence, a multimodal, noninvasive, and comprehensive evaluation employing biochemical, molecular, cellular, and high-throughput system biology techniques are required to respond to the rising burden of CRC. Using a high-throughput quantitative tissue proteomics technique, this study aimed to unravel the proteome dysregulation in CRC tissue samples.
The signaling pathways necessary for homeostasis maintenance are often abnormally reactivated during carcinogenesis, giving cells an edge in proliferation, a better chance of surviving, and invasion ability. Moreover, cancer cells require enormous quantities of biomolecules, adenosine triphosphate, and nucleotides to maintain rapid proliferation. To match the required potential, tumor cells often exhibit an increased flux of glycolysis through central carbon metabolism (Martínez-Reyes and Chandel, 2021). Tumors activate pyruvate metabolism, which transforms the unused pyruvate into lactate to continue the glycolytic flux. Along with the energy metabolism, structural and functional requirements were served through the reorganization and hyperactivation of multiple biological cascades to achieve immortalization.
During the process, the transformed cell enhances its in-house manufacturing units such as remodeling of the chromatin, purine metabolism, transcription machinery, spliceosomes, mRNA transports, translation units, and cargo trafficking across the cellular architecture (Hanahan and Weinberg, 2011).
All these processes cumulatively lead to the cytoskeleton remodeling, activation of the ion channels, reorganization of the cell–cell and cell–extracellular matrix (ECM) junctions, etc. making a favorable environment for cancer to proliferate extensively.
An analogous biological event was identified in the study where the high-resolution quantitative proteomics-based experiment was done to understand the molecular insight into CRC pathobiology. In the present study, a series of proteins were identified to be highly perturbed, which helped in understanding the disease more clearly. Keeping the diagnostic potential of these proteins targeted proteomics-based validation was carried out to gain more confidence. A list of proteins, including GRAN, ANPEP, YY1, OLFM4, NUDT2, PADI2, and MMP9, etc. was identified to be elevated in the disease, which further validated to understand the disease precisely.
GRAN a calcium-binding protein, aids in neutrophils' adherence to fibronectin. Although several studies have established the proinflammatory role of the protein, its role in cancer development is still not explored. Recent studies have established its role in NFkB activation, and MAPK activation in dendritic cells (Kim et al, 2016). The quantitative analysis of the tumor has shown GRAN to be highly upregulated, which is further identified from the logistic regression analysis and validated using targeted proteomics showing the protein as an excellent candidate for disease diagnosis.
YY1, the versatile transcription factor controls the transcriptional regulation of numerous genes involved in a variety of cellular activities, including cell proliferation, the balance of death and survival, DNA repair, chromatin remodeling, autophagy, etc. YY1 has been identified as a key player in the development of several malignancies due to its function in pathways that, when dysregulated, are connected to malignant transformation. The protein is identified to have both oncogenic as well as a tumor suppressor in cancers. The upregulation in the protein is reported in multiple cancers, including breast, lung, prostate, cervical cancer, etc., and the tumor suppressor role of YY1 is well established in pancreatic cancer (Sarvagalla et al, 2019). In CRC, YY1 has primarily been demonstrated to be protumorigenic. O-GlcNAcylation has recently been demonstrated to have a significant role in modifying protein stability and function, exhibiting essential oncogenic properties (Zhu et al, 2019). OLFM4 is an antiapoptotic ECM glycoprotein that interacts with cadherins and lectins and facilitates cell adhesion (Ohkuma et al, 2020; Quesada-Calvo et al, 2017, p. 4). The protein is established as an intestinal stem cell marker and identified to be heavily expressed in CRC.
Recent studies have reported an association of OLFM4 with multiple cancers, including gastric, pancreatic, colon, lung, and breast cancer, and so on. The protein is also identified in liver metastases of cervical and CRC origin. Depletion of the protein in gastric cancer cells has also been shown to inhibit cell growth and proliferation and apoptosis induction through TNFα signaling cascade (Liu et al, 2012, p. 4). The current study shows upregulation of OLFM4 and validates the findings of earlier research and increases confidence in the protein's ability to serve as a diagnostic marker for CRC. AMPN, a metallopeptidase, is essential for the subsequent digestion of peptides generated from the proteolysis of gastric and pancreatic proteases (Sørensen et al, 2013). Besides, this protein is also identified to be involved in the processing of peptide hormones, neuropeptides, and chemokines. AMPN is reported to be highly elevated in breast, ovarian, thyroid, colon cancer, etc. (Wickström et al, 2011). In renal cancer, the enzyme is identified to be downregulated (Blanco et al, 2014).
The enzyme is already recognized as a marker for angiogenesis, and it is only necessary for angiogenic endothelial cells to function but not for regular vasculature. Also, the enzyme takes a definite part in endocytosis, signal transduction, and plays a crucial role in cell migration, invasion, angiogenesis, and metastasis. (Li et al, 2020). In the present study, the protein is identified to be significantly elevated showing the role of the ectoenzyme in the development and metastasis of CRC.
The use of matched tumor and control tissues is the primary strength of the current investigation. By doing this, the variance between individuals brought on by their genetic make-up, way of life, and environmental factors was eliminated. Despite the biological differences among the samples, the expression levels in the proteins were identified to be consistent. A significant drawback of the current study is that both discovery and validation were carried out on a small sample cohort due to the lack of access to a large number of clinical samples. Despite using tumor specimens from patients with different ethnic origins, the changes in the protein expression were consistent among the CRC patients. Due to the unavailability of a large number of clinical samples, both discovery and validation were performed on a small sample cohort, which is a major limitation of the present study. Synthetic peptide usage in the MRM experiments aids in the accurate quantification of peptides.
MRM assays reported in the study were performed without synthetic peptides due to their unavailability; hence, the relative abundance of the peptides for the significantly altered proteins was measured, instead of absolute quantification, which is the second limitation of the study. Significantly altered proteins discovered from the current investigation can be further confirmed in bigger patient cohorts.
Conclusions
We identified several significantly altered proteins, which we suggest play potentially key roles in the pathophysiology of CRC. Various transcriptional factors were found to be associated with the altered proteins. Importantly, a candidate panel of proteins, including GRAN, ANPEP, OLFM4, among others, showed promise toward future biomarker development to inform CRC diagnosis. We call for future translational research to evaluate and validate these proteins on larger independent patient cohorts in India and other world populations.
Footnotes
Authors' Contributions
A.B., C.K., and S.S. conceived and designed the project. A.B. collected and prepared samples, and optimized and collected spectral acquisition. A.H. and S.P. did the data analysis and GSEA analysis. A.D. did the biological pathway analysis. A.B. drafted the article. S.S., C.K., A.H., A.D., and S.P. edited the article. All authors made a significant intellectual contribution and have approved the final version of the article.
Author Disclosure Statement
The study was funded in part by grants from the UGC and Merck Center of Excellence to S.S. The funders had no role in the design, execution, analysis, writing, or reporting of the present study. The authors declare they have no financial conflicts of interest.
Funding Information
We would like to acknowledge UGC for funding this work (Project code: F.6-17/2014) and also acknowledge Merck Center of Excellence (DO/2021-MLSP) granted to S.S. A.B. is supported by a CSIR fellowship in India for Ph.D. We are thankful to the MASSFIITB facility for PRM and quantitative proteomic analysis.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.