
Abstract
COVID-19 caused by the SARS-CoV-2 infection is a systemic disease that affects multiple organs, biological pathways, and cell types. A systems biology approach would benefit the study of COVID-19 in the pandemic as well as the endemic state. Notably, patients with COVID-19 have dysbiosis of lung microbiota whose functional relevance to the host is largely unknown. We carried out a systems biology investigation of the impact of lung microbiome-derived metabolites on host immune system during COVID-19. RNAseq was performed to identify the host-specific pro- and anti-inflammatory differentially expressed genes (DEGs) in bronchial epithelium and alveolar cells during SARS-CoV-2 infection. The overlapping DEGs were harnessed to construct an immune network while their key transcriptional regulator was deciphered. We identified 68 overlapping genes from both cell types to construct the immune network, and Signal Transducer and Activator of Transcription 3 (STAT3) was found to regulate the majority of the network proteins. Furthermore, thymidine diphosphate produced from the lung microbiome had the highest affinity with STAT3 (−6.349 kcal/mol) than the known STAT3 inhibitors (n = 410), with an affinity ranging from −5.39 to 1.31 kcal/mol. In addition, the molecular dynamic studies showed distinguishable changes in the behavior of the STAT3 complex when compared with free STAT3. Overall, our results provide new observations on the importance of lung microbiome metabolites that regulate the host immune system in patients with COVID-19, and may open up new avenues for preventive medicine and therapeutics innovation.
Introduction
COVID-19 is a highly contagious disease caused by the SARS-CoV-2 virus (Sadeghi Dousari et al., 2020). SARS-CoV-2 is an enveloped virus (Pal et al., 2020) that produces a broad range of cellular changes and clinical symptoms in multiple tissues that can be further compounded by comorbid diseases such as hypertension, type 2 diabetes, and cardiovascular diseases (Ejaz et al., 2020). Because COVID-19 is a systemic disease, a systems biology approach would be well poised to study its pathophysiology.
Angiotensin-converting enzyme 2 receptor is the entry point of SARS-CoV-2 into the host, which gets replicated at the lower respiratory tract, causing inflammatory cytokine storms, pneumonia, and multiorgan failure (Leung and Wong, 2020). Acute lung injury and respiratory distress syndrome are notable causes of COVID-19 mortality (Aslan et al., 2014). With infection, the lung gene expression changes to activate multiple immune signals to release large numbers of intracellular inflammatory factors, including interleukin 6 (IL-6), interleukin 1 beta (IL-1β), tumor necrosis factor alpha (TNF-α), interferon (IFN), and complement protein (Paudel et al., 2020). Interestingly, and in a larger integrative biology context, studies in the past suggested that host microbiome can play an essential role in regulating immune signals against viral infection (Belkaid and Hand, 2014).
Recent technologies have demonstrated the importance of host microbiota in regulating variety of cellular function (Paudel et al., 2020). Approximately 10–100 trillion microbes were reported to be a part of the human system, which includes bacteria, viruses, and fungi. Among these microbial families, bacteria were observed to be more prominent, covering almost 2000 different species (Ursell et al., 2012). These microbes have a significant impact on host immune activation and function. Such mechanisms are achieved through the direct interaction of the microbiome or by its producing metabolites with the host immune system to prevent serious infection (Belkaid and Hand, 2014). Maintaining lung homeostasis and accelerating adequate immune responses against pathogens are two specific tasks of lung microbiome (Sommariva et al., 2020).
However, very limited studies are available on the influence of lung microbiota in patients with COVID-19 and its impact on host immune response. With the advent of recent omics technologies, the systems biology concept of integrating various high-throughput omics data has become possible to predict or assess microbial virulence, molecular pathogenesis, biomarker discovery, and potential drug targets (Jaiswal et al., 2020). Currently, the systems biology approach has broadened our understanding of COVID-19, opening the door to new drug targets and treatment strategies. For example, Fava et al. (2022) used a systems biology approach to identify candidate drugs to reduce COVID-19 mortality. Likewise, Hasan et al. (2022) identified the crucial blood-based signature molecules in relation to biomarkers and drug targets in patients with COVID-19.
Herein, we applied systems biology approach (Fig. 1) that aimed to assess the effects of lung microbiome-derived metabolites in regulating the altered immune protein networks in COVID-19. Such investigation could help to identify crucial metabolites produced by the microbiome that can effectively shield the host from SARS-CoV-2. Our assessment integrates series of computational methods, including RNAseq analysis, protein interaction network, molecular docking, and molecular dynamic (MD) simulation. Using these methods, we evaluated the RNAseq data set to identify the differentially expressed genes (DEGs) of lungs infected with SARS-CoV-2.
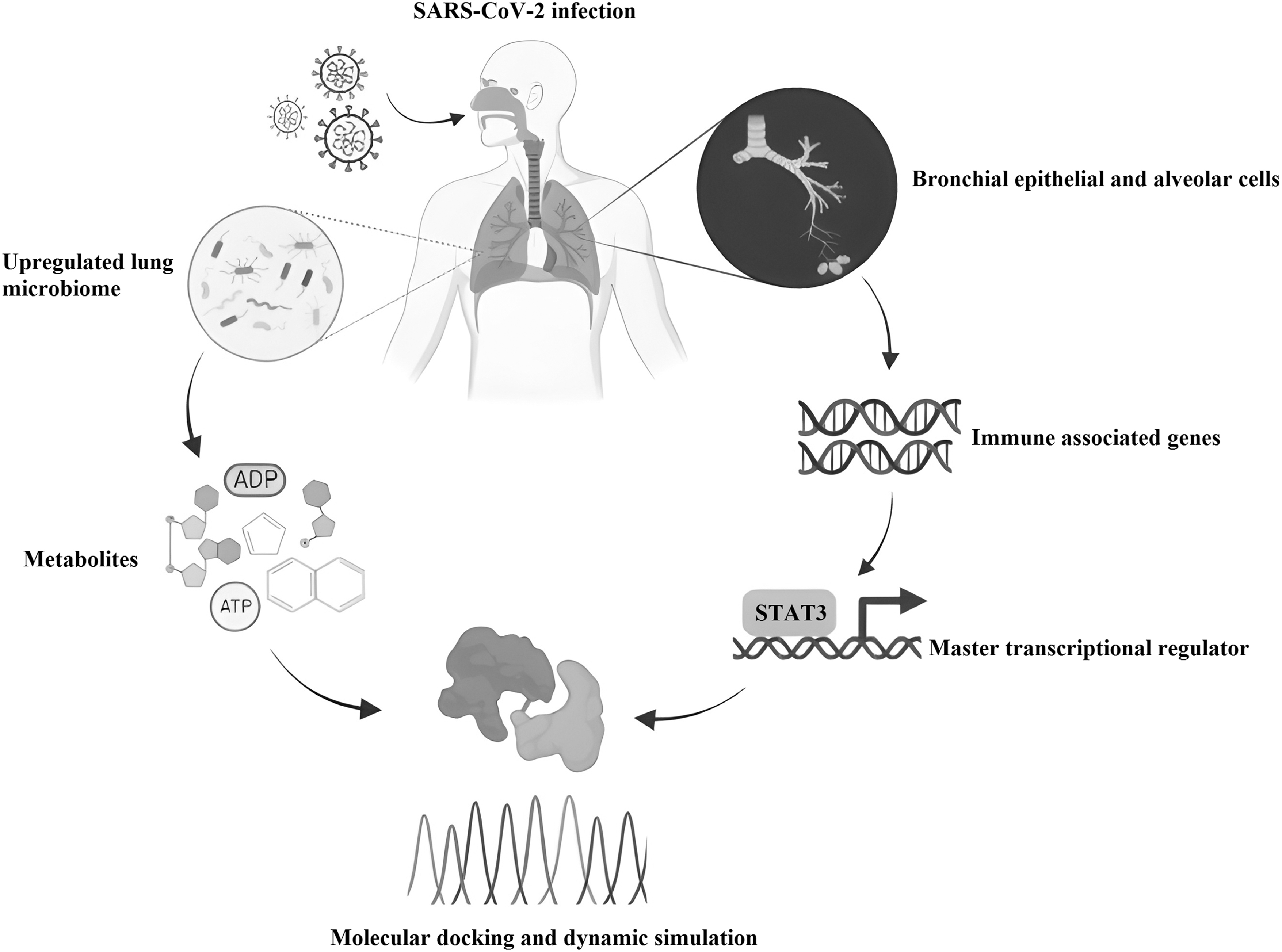
Graphical summary representing the workflow of our approach. ADP, adenosine diphosphate; ATP, adenosine triphosphate; STAT3, signal transducer and activator of transcription 3.
Then, we constructed the immune protein network from the DEGs, which demonstrates the behavior of pro- and anti-inflammatory molecules in COVID-19. Furthermore, we established the key transcriptional regulators of the immune network that is modulated by the metabolites of lung microbiome. Overall, our research points to the importance of metabolites from lung microbiota to modulate the key regulators of pro- and anti-inflammatory molecules to potentially protect the host during COVID-19.
Materials and Methods
Mapping and analyzing DEGs
The publicly available and relevant data sets containing the transcriptome profile of SARS-CoV-2-infected lung cells were searched in the NCBI Gene Expression Omnibus (GEO data sets) repository. This study was conducted under the overall research ethics oversight of the authors' institution. Based on the following criteria, the lung transcriptome profile was chosen from a variety of data sets: (1) expression profiling performed using high-throughput sequencing (RNAseq), (2) expression across different lung cells on SARS-CoV-2 infection, (3) minimum samples (n ≥ 3) per group for case and control under single controlled experiment, and (4) adequate sample and platform information for the subsequent analysis.
Accordingly, the GSE147507 that is consisted of nine cases and nine controls were chosen to retrieve the raw files in FASTQ format. The RNA-seq workflow was implemented that included (A) FASTQC tool to ensure high-quality reads, (B) STAR algorithm to align the qualified reads to the human genome (hg 19), and finally (C) DESeq2 to identify DEGs (log2FC) with 1.5-fold change and p ≤ 0.05 in each SARS-CoV-2-infected cells compared with its control.
Gene enrichment and network construction
The shared DEGs of both bronchial epithelium and alveolar cells were used to perform functional analysis using Gene Ontology (GO) database (www.informatics.jax.org). From the functional assessment, we selected DEGs that exhibit pro-inflammatory (GO: 0050729) and anti-inflammatory (GO: 0031348) properties. Furthermore, the selected pro- and anti-inflammatory genes were assumed as proteins and subjected to the GeneMANIA (Gene Multiple Association Network Integration Algorithm) (Zhong et al., 2021) plug-in of Cytoscape 3.9.1 version (Shannon et al., 2003) to construct the immune network.
Transcriptional regulatory network
The Cytoscape version 3.9.1 iRegulon (Janky et al., 2014) plug-in was used to identify the master transcriptional regulators of the immune network. The iRegulon employs a genome-wide ranking technique to discover transcription factor (TF) motifs that are enriched for the given specified protein targets. Hence, the immune network proteins were inputted to discover their TFs. Among multiple TFs, the TF that regulates most of the immune network proteins was selected.
Lung microbiota-derived metabolites
Concurrently, a search of the literature was conducted in NCBI, PubMed database to discover the relevant publication describing the microbiota of bronchoalveolar lavage fluid (BALF) in SARS-CoV-2-infected patients. Among the search results, Han et al. (2022) was noticed useful that presents a landscape of BALF microbiota in 23 controls and 19 SARS-CoV-2-infected cases from six studies using meta-transcriptomics. From the supplementary file of Han et al. (2022), the over-represented microbes in BALF across all six studies were selected. The list of metabolites produced by these over-represented bacteria were subsequently retrieved from the Virtual Metabolic Human (VMH) database (https://www.vmh.life/). Then, the structure of each metabolite was extracted from the NCBI, PubChem database in structure-data file format for subsequent analysis.
Molecular docking
Using the glide docking module in the Schrodinger software (Glide; Schrodinger, LLC, New York, NY, 2021), the binding efficiency of each of the collected metabolite with a TF protein was examined. The structure of TF protein was retrieved from the Protein Data Bank (PDB ID: 6QHD) and preprocessed by the Protein Preparation Wizard in Maestro 11.2 of Schrodinger (Protein Preparation Wizard; Epik; Schrodinger, LLC, 2021).
The active site of protein was identified from P2Rank server (Krivák and Hosza, 2018) and grid box was generated using the receptor grid construction tool in the Maestro 11.2. In contrast, each collected metabolite structure was optimized with LigPrep in Maestro 11.2 (LigPrep; Schrodinger, LLC, 2021). Glide dock module from Schrodinger suite was used for molecular docking. All docking calculations were performed using the OPLS3 force field in SP (Standard Precision) mode. In addition to microbial metabolites, the known TF protein inhibitors were retrieved from CHEMBL and their binding efficiencies were evaluated based on the least energy confirmation.
MD simulation
After the docking study, the efficiently bounded microbial metabolite with the TF protein was taken for MD simulation. The GROMACS 2021.6 version (Abraham et al., 2015) was employed to perform simulations with the CHARMM-27 force field and TIP3P water model. The SwissParam tool was used to create the ligand topology file. The complex was simulated inside the cubic box with ions neutralized by adding Na+ and Cl–. Energy minimization was performed to avoid poor contacts and clashes in the complex using the steepest decent approach. After energy minimization, constant number, volume, temperature (NVT) and constant temperature, constant pressure ensemble (NPT) equilibration were carried out. The simulations were conducted for 100 ns to evaluate the trajectories based on root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA), and hydrogen bonds (HBs).
Results
Mapping and analyzing DEGs
Appropriate transcriptome data set (Accession ID: GSE147507) was selected from NCBI, GEO data sets based on the selection criteria. Further to identify DEGs, the raw FASTQ data of control and SARS-CoV-2-infected bronchial epithelial and alveolar cells were analyzed separately using the RNAseq pipeline as mentioned in the methodology section. Notably, 1305 genes were upregulated and 1169 were downregulated in bronchial epithelium, whereas in alveoli 4723 were upregulated and 4372 were downregulated in SARS-CoV-2 infection.
Immune protein network and regulators
Among these DEGs, 1767 genes were shared between bronchial epithelial and alveolar cells. Based on the gene ontology analysis, 68 genes out of 1767 were associated with either pro- or anti-inflammatory properties (Supplementary Table S1). These 68 genes encoded proteins were utilized in the construction of an immune network (Fig. 2). Furthermore, iRegulon was used to identify the TFs that are responsible for regulating the proteins found in the network. Notably, we found that the STAT3 was the key TF regulating the more number (n = 47) of proteins (Fig. 3). As a direct consequence of this, STAT3 was selected as a potential target that could be regulated by microbial metabolites.

Immune protein network—Represents the network of 77 pro- and anti-inflammatory genes that are commonly found in bronchial epithelia and alveolar tissues.
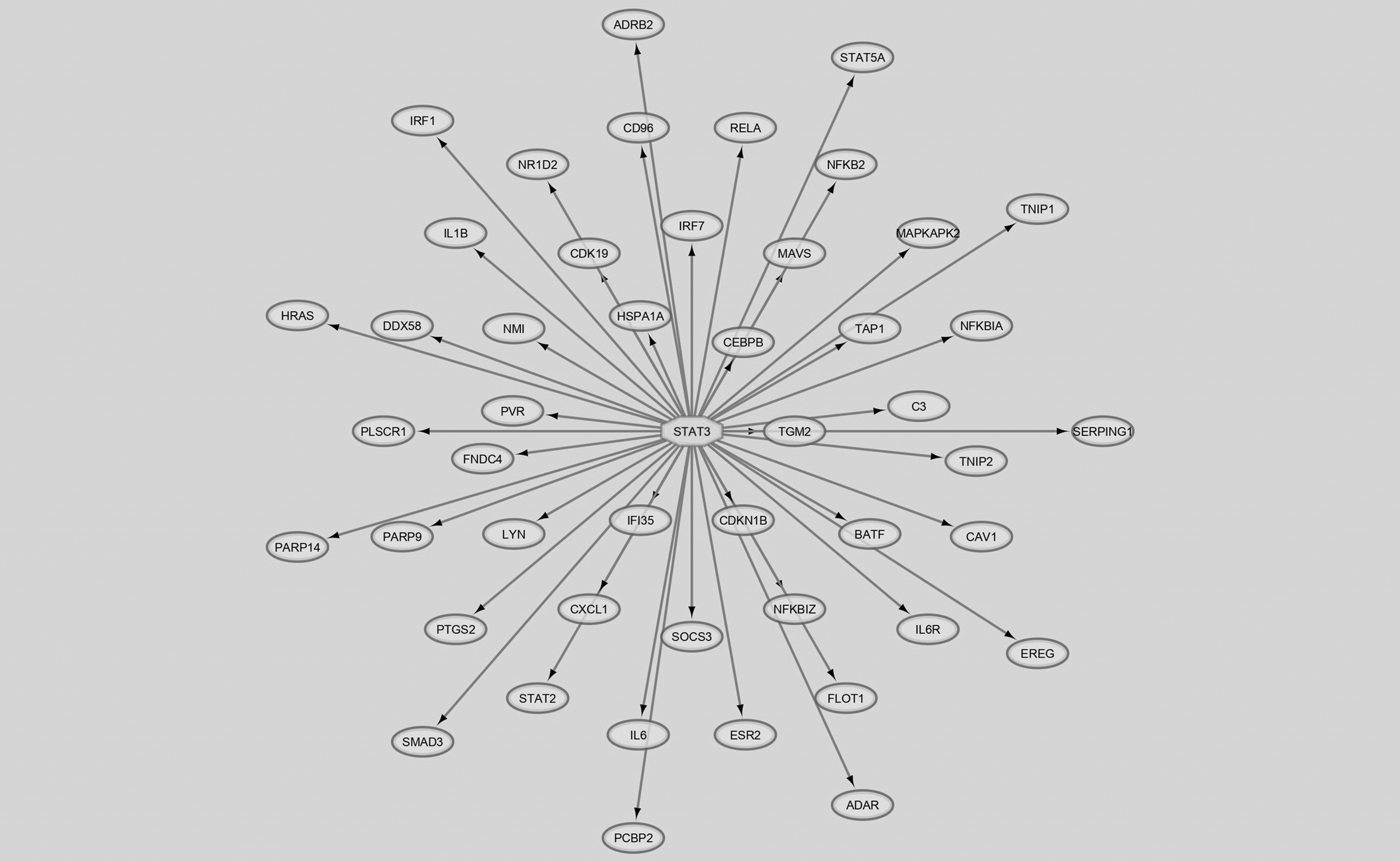
Regulatory network containing 47 proteins regulated by the STAT3 transcription factor.
Lung microbiota-derived metabolites
On the basis of supplementary information provided by Han et al. (2022), a list of 95 over-represented bacterial species of BALF in SARS-CoV-2 infection were selected (Supplementary Table S2). Furthermore, the metabolites (n = 1231) produced by these microbes were collected from the VMH database, and their structures were downloaded from the PubChem databases. In addition, the known STAT3 inhibitors (n = 410) were retrieved from CHEMBL database to perform molecular docking.
Docking and dynamic simulation of STAT3 with microbial inhibitors
The binding affinity of known inhibitors (n = 410), as well as microbial metabolites (n = 1231), was investigated against STAT3 protein (Figs. 4A–D and 5A). P2Rank was used to locate the grid box that encompasses the active site residues (THR620, SER613, GLU612, SER611, ARG609, LYS591, PRO639, GLU638, VAL637, SER636, and GLN635) of STAT3. The binding energies of known STAT3 inhibitors were ranged around −5.39 to 1.31 kcal/mol (Supplementary Table S3) (Fig. 4B–D). Whereas among the microbial metabolites, the thymidine diphosphate exhibited the highest affinity toward STAT3 with lowest binding energy −6.349 kcal/mol (Figs. 4A and 5A).

Two-dimensional interaction plots representing the STAT3 with

Interaction diagram of STAT3 and thymidine diphosphate
As a consequence of inhibition, it can modulate the immune network of the lungs, and thereby protect the host from inflammation. Further the MD simulations were performed, to determine the stability of free STAT3 and STAT3 complex (STAT3 with thymidine diphosphate). Comparative RMSD analysis demonstrate the stability of STAT3 protein on thymidine diphosphate binding by exhibiting less deviation from the ligand-free STAT3 (Fig. 5B). In addition, the RMSF plot revealed a shift in the fluctuation of particular residues involved in thymidine diphosphate binding (Fig. 5C). Similar differences in the trajectory of Rg, SASA, and HBs were observed between the free and STAT3 complex (Fig. 5D–F).
Discussion
This study highlights the importance of metabolites from lung microbiota to modulate the key regulators of pro- and anti-inflammatory molecules to potentially protect the host during COVID-19. It also attests to the value of systems biology approaches to unravel the complex pathophysiology of COVID-19 with an eye to develop preventive medicine and therapeutics innovations. Systems biology has been shown to be valuable in various complex disorders such as cancer, diabetes (Somvanshi and Venkatesh, 2014), neurological (Savelieff et al., 2022), and autoimmune diseases (Kim et al., 2014). However, there is a paucity of studies on the microbiota of COVID-19-infected individuals. Interestingly, Johnson et al. (2019) explored the importance of bacterial surface component (peptidoglycans) on COVID-19 severity. Also, Hernández-Terán et al. (2021) reported the dysbiosis of lung microbiome and its potential impact on the clinical outcomes.
In this study, we compared the transcriptomic profiles of bronchial epithelium and alveolar cells in SARS-CoV-2-infected and healthy controls to determine the DEGs. The selected DEGs with log2FC and p < 0.05, resulted in 1761 common genes between the bronchial epithelium and alveolar cells. These common genes were further analyzed for their molecular function by gene ontology. Of the 1761 genes, 68 (44 pro- and 24 anti-inflammatory) were associated with immunological function (Supplementary Table S1).
All 68 immune-related genes were then employed to construct the immune protein network. Notably, the network construction tool GeneMANIA expanded our query list and displayed an interactive immune network containing 77 proteins (Fig. 2). Using iRegulon plug-in, STAT3 was identified as the master transcriptional regulator of immune network that regulates a maximum of 47 genes (Fig. 3). Whereas nuclear factor beta (NF-B) regulates 46 genes, STAT2 and CCAAT enhancer binding protein (CEBP) individually regulate 33 genes in the network. Jafarzadeh et al. (2021) describe the role of STAT3 in initiating inflammation and suppressing the interferon response during COVID-19.
Also, STAT3 elevates the disease severity through M2 macrophage polarization, fibrosis, thrombosis, and a cytokine storm (Jafarzadeh et al., 2021). Therefore, targeting the STAT3 TF that regulates various immune-related genes could potentially benefit the patient with COVID-19. Particularly, Belkaid and Hand (2014) suggest that the host microbiome and their metabolites can play significant role in regulating immune system against viral infection. Hence, we tested 1231 microbial metabolites of over-represented bacterial species as reported by Han et al. (2022) (Supplementary Table S2) against STAT3 through molecular docking.
As a result of molecular docking, thymidine diphosphate showed highest affinity against STAT3 with the lowest energy value of −6.349 kcal/mol (Supplementary Table S3). Thymidine diphosphate makes HBs to inhibit STAT3 at residues GLU612, ARG609, LYS591, THR620, VAL637, and SER636 (Fig. 4A). From the Supplementary Table S4, thymidine diphosphate metabolite was found to be secreted by more than 52 over-represented bacterial species, including Lactobacillus and Pseudomonas genus. We also performed docking with 410 known STAT3 inhibitors from the CHEMBL database for comparative purposes. These compounds (Fig. 4B–D) exhibited binding energies between −5.399 and −1.31 kcal/mol (Supplementary Table S3). Hence, thymidine diphosphate has the highest affinity of all known STAT3 inhibitors.
MD simulation was performed to determine the dynamic behavior of free STAT3 and STAT3 complex. Each system underwent a simulation period for 100 ns, and their trajectories were compared. RMSD analysis showed binding of thymidine diphosphate to the active site of STAT3 generated less structural and conformation variation when compared with free STAT3 (Fig. 5B). However, a few minor fluctuations were observed between 15–20 ns (0.34 nm) and 75–80 ns (0.42 nm), but stable equilibrium was maintained. Likewise, RMSF (Fig. 5C) showed residual fluctuations at distinct regions when thymidine diphosphate binds to STAT3. Yet, these fluctuations do not influence the total structural stability of STAT3.
We then computed the protein conformational stability in a biological system using the Rg. The greater flexible protein packing confers a greater Rg value. The average Rg values of free and STAT3 complex were 3.614 and 3.671 nm, respectively (Fig. 5D). The Rg plot showed high compactness after the binding of thymidine diphosphate to STAT3. We also employed SASA, to evaluate the STAT3 interaction with surrounding solvent. The average SASA values of free and STAT3 complex were 311.981 and 325.318 nm2, respectively (Fig. 5E). Higher SASA values of the complex indicates conformational change that exposes few STAT3 residues to the surrounding solvent.
The intermolecular H-bond interactions were also determined to analyze the binding strength of thymidine diphosphate to the active sites of STAT3 (Fig. 5F). Thymidine diphosphate with STAT3 establishes an average of 427 HBs, which indicates its stronger binding potency. Our results sparked speculation that STAT3 inhibition by lung microbiota-derived metabolites could possibly affect the regulation of the immune network to protect the host from SARS-CoV-2.
Conclusions
This study with a systems biology approach lend evidence for the impact of lung microbiome-derived metabolites on host immune system in COVID-19. Particularly, molecular docking and dynamic simulation revealed that thymidine diphosphate metabolite contributed by lung microbiome can significantly inhibit STAT3 TF. Such inhibition regulates pro- and anti-inflammatory molecules during COVID-19. Further in silico and clinical research are warranted in regard to the role of the lung microbiome-derived metabolites in a context of preventive medicine, and drug discovery and development.
Footnotes
Acknowledgments
The authors thank their institutes for research support and encouragement.
Authors' Contributions
Conceptualization and supervision by S.S.S.J.A. Experiments by J.R., M.S., S.B., F.A.B., S.P., M.K., and A.A. Data analysis, writing, and editing the article by J.R. and M.S.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This project was funded by the Researchers Supporting Project no. RSPD 2023R653, King Saud University, Riyadh, Saudi Arabia.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.