
Abstract
The Hippo signaling pathway is a master regulator of development, cell proliferation, and apoptosis in particular, and it plays an important role in tissue regeneration, controlling organ size, and cancer suppression. Dysregulation of the Hippo signaling pathway has been implicated in breast cancer, a highly prevalent cancer affecting 1 out of every 15 women worldwide. While the Hippo signaling pathway inhibitors are available, they are suboptimal, for example, due to chemoresistance, mutation, and signal leakage. Inadequate knowledge about the Hippo pathway connections and their regulators limits our ability to uncover novel molecular targets for drug development. We report here novel microRNA (miRNA)-gene and protein–protein interaction networks in the Hippo signaling pathway. We employed the GSE miRNA dataset for the present study. The GSE57897 dataset was normalized and searched for differentially expressed miRNAs, and their targets were searched using the miRWalk2.0 tool. From the upregulated miRNAs, we observed that the hsa-miR-205-5p forms the biggest cluster and targets four genes involved in the Hippo signaling pathway. Interestingly, we found a novel connection between two Hippo signaling pathway proteins, angiomotin (AMOT) and mothers against decapentaplegic homolog 4 (SMAD4). From the downregulated miRNAs, hsa-miR-16-5p, hsa-miR-7g-5p, hsa-miR-141-3p, hsa-miR-103a-3p, hsa-miR-21-5p, and hsa-miR-200c-3p, target genes were present in the pathway. We found that PTEN, EP300, and BTRC were important cancer-inhibiting proteins, form hubs, and their genes interact with downregulating miRNAs. We suggest that targeting proteins from these newly unraveled networks in the Hippo signaling pathway and further research on the interaction of hub-forming cancer-inhibiting proteins can open up new avenues for next-generation breast cancer therapeutics.
Introduction
Breast cancer is one of the most common cancers worldwide (Verma et al., 2019), and it is affecting 1 out of every 15 women worldwide. In the majority of cases, dysregulation of cell growth pathways is observed. The Hippo signaling pathway, which is conserved from protista to eukaryotes (Yang and Hata, 2013), is a master regulator of development, cell proliferation, stem cell function, tissue regeneration, homeostasis, and organ size, and cancer suppression. However, dysregulation promotes cancer cell growth, invasion, migration, and resistance to chemotherapy (Mohajan et al., 2021).
Numerous studies have shown the connection between the overexpressed Hippo pathway's key proteins and cancer cell proliferation, invasion, migration, and drug resistance (Zanconato et al., 2016). For example, overexpression of YAP leads to diverse cancers (Camargo et al., 2007; Dong et al., 2007; Lee et al., 2008). YAP/TAZ overexpression is common in breast cancer (Diaz-Martin et al., 2015; Maugeri-Sacca et al., 2015). In highly invasive and chemoresistant breast cancer, TAZ is overexpressed (Bendinelli et al., 2013; Xiang et al., 2015). Under some conditions, blocking the overexpressed proteins of an important dysregulated pathway is beneficial in preventing cancer growth.
Besides chemoresistance, inadequate knowledge about the pathway connections and their regulators limits our ability to uncover novel targets. A deeper knowledge of the Hippo pathway can aid in developing effective anticancer and anti-chemo resistance medications (Huang et al., 2005). Recent in vivo and in vitro studies have revealed that microRNAs (miRNAs) can target and repress various cancer pathways. The use of miRNAs in treatments that target cancer pathways is more cell-friendly than standard therapies such as radiation, chemotherapy, and surgical intervention, and with fewer adverse effects (Astamal et al., 2020).
The miRNAs are tiny, endogenously produced, non-coding RNAs that target gene expression to govern cellular activities, including development, differentiation, apoptosis, and proliferation (Chen and Rajewsky, 2007; Krol et al., 2010). Dysregulated miRNAs influence many transcripts and significantly impact cancer-related signaling pathways (Sand et al., 2012). Several miRNAs, called “oncomiRs,” act as oncogenes and create an oncogenic phenotype (Mittal et al., 2021).
Targeting miRNA to inhibit the progression of many illnesses, particularly breast cancer, is gaining momentum nowadays. miR-21, miR-10b, miR-221/222, and miR-155 genes are overexpressed and promote breast cancer; inhibiting them showed significant results against breast cancer (Guo et al., 2018; Kong et al., 2013; Stinson et al., 2011; Yan et al., 2008). On the other hand, several operate as tumor suppressors and limit cancer progression (Astamal et al., 2020).
miR-34a is typically downregulated in breast cancer cells, and delivering molecules that mimic it can restore its expression, and inhibit the expression of target oncogenes (Rui et al., 2018; Xiao et al., 2016; Zhao et al., 2013). Targeting a single miRNA will not help much, however, and targeting an overexpressed cluster using miRNA inhibitors and miRNA mimics will (Kabekkodu et al., 2020; Wang and Wu, 2009; Yoshida et al., 2021). As a result, identifying more miRNAs through network biology can aid next-generation drug development (Astamal et al., 2020; Kandettu et al., 2020).
The aim of the present study was to uncover miRNA-gene and protein–protein interaction networks in the mammalian Hippo signaling pathway using in-silico analyses (Fig. 1). Further, we investigated the potential roles of the proteins using functional and pathway enrichment analysis, with an eye to therapeutics discovery and development for breast cancer.
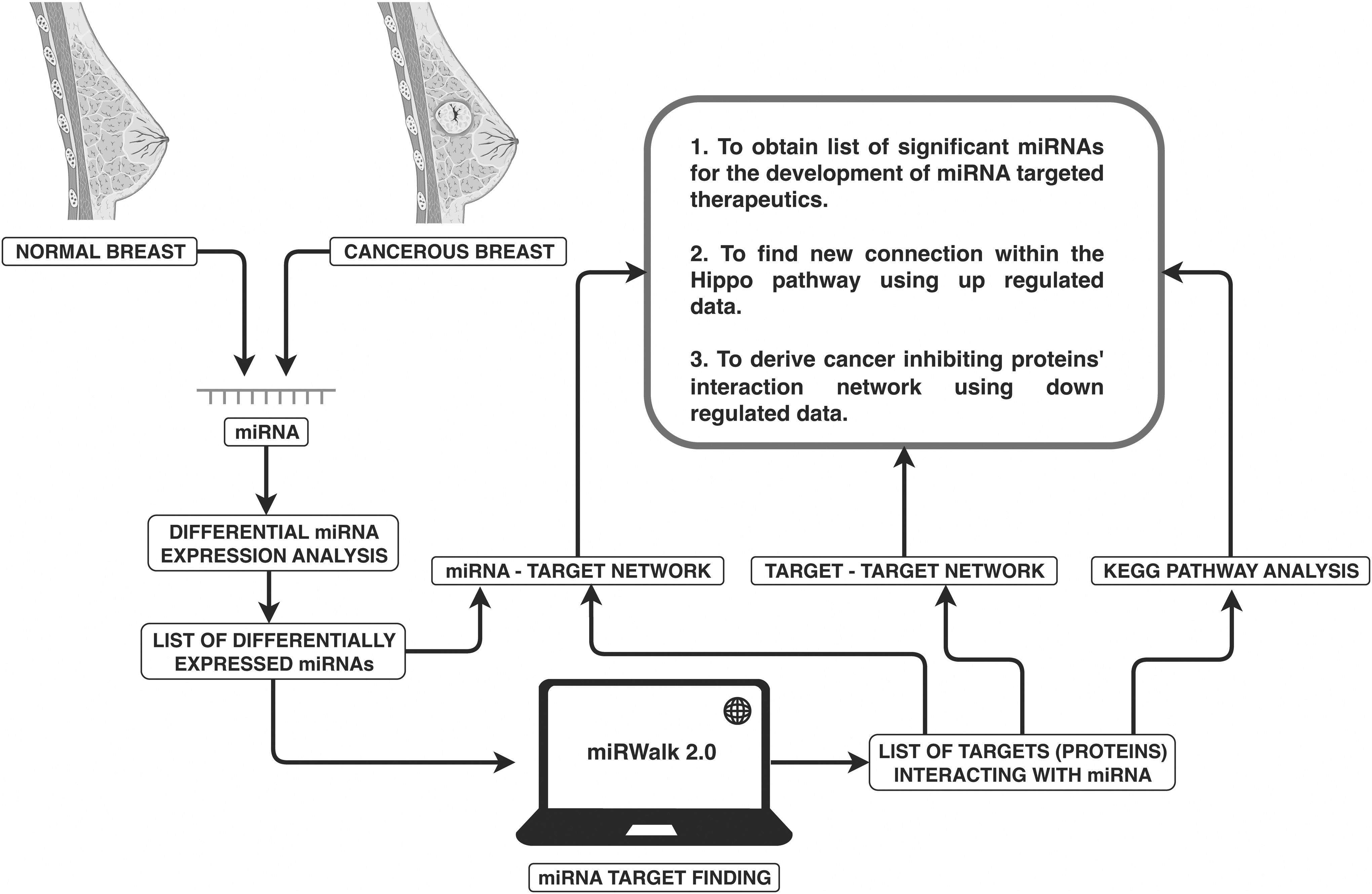
Study design. The GSE57897 dataset with cancerous and non-cancerous data was used to identify the differentially expressed miRNAs. The list of miRNAs was uploaded to identify the target proteins. These proteins were further analyzed to determine a novel link in the Hippo signaling pathway. miRNAs, microRNAs.
Materials and Methods
Differential analysis
We used the GSE57897 dataset, having 31 non-cancerous and 422 cancerous samples, to perform differential miRNA expression analysis (Zhao et al., 2019). Data were collected from patients who had undergone curative mastectomy between 1999 and 2006. Aside from the sample size and timeframe, the dataset distinguishes invasive ductal carcinoma from non-cancerous breast that aligns with the purpose of the study.
Before performing the t-test, NCBI's GEO2R tool is used to normalize the data. The same tool distinguishes between differentially and non-differentially expressed miRNAs. For further study, we selected only those miRNAs with an adjusted p-value >0.05 and a Log2 value greater than 1. For volcano plot, p-value is converted into a -log10 value and loaded in the “VolcaNoseR” web program (https://huygens.science.uva.nl/VolcaNoseR/) (Goedhart and Luijsterburg, 2020). This study used publicly accessible data and did not involve the recruitment of human subjects or animal research. The study was conducted under the overall research ethics oversight of the authors' institution.
Target gene identification
We used the miRWalk2.0 tool (http://mirwalk.umm.uni-heidelberg.de) to find the genes targeted by miRNAs (Sticht et al., 2018). In addition to its database, the tool searches two other well-known databases, TargetScan and miRDB. We selected only genes that are validated and present in all three databases.
Functional enrichment analysis
Functional enrichment studies were conducted to understand these targeted genes' function better. This approach finds biological pathways more enriched in a gene list than expected by chance. We used DAVID and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses (Jiao et al., 2012). DAVID is a free online resource that provides a functional interpretation, focusing on Gene Ontology (GO) keywords (Tomczak et al., 2018).
The GO enrichment analysis comprises biological processes, cellular components (CC), and molecular functions (MF), whereas KEGG analysis is a set of operations that provide an understanding of high-level functions and utilities of the biological system (Kanehisa and Goto, 2000).
Networking
We used Cytoscape to create miRNA-gene and protein–protein networks (Shannon et al., 2003). Within the Cytoscape, the STRING app uploads the list of proteins to create protein–protein interaction networks (Szklarczyk et al., 2019). The application automatically retrieves the interaction networks and displays them in Cytoscape's working area for further analysis. On the other hand, the KEGG database extracted the list of proteins involved in the Hippo signaling pathway. Both lists were compared to find the common proteins highlighted in the network. The connections generated by the highlighted proteins in the networks were given special attention.
Clustering
We used the ClusterMaker2 app of the Cytoscape tool to divide large networks into their meaningful clusters (Morris et al., 2011). To find clusters, we used the Markor Cluster Algorithm (MCL) based on the random walk principle with iteration 4. Clustering aims at dividing the network into groups with similar characteristics. It aids in the identification of key nodes and their relationships with other nodes (Enright et al., 2002; van Dongen and Abreu-Goodger, 2012).
Results
Out of the 53 upregulated miRNAs, we found 6 miRNAs to have validated targets, whereas, from 49 downregulated miRNAs, we found 9 miRNAs to have validated targets (Fig. 2a). The 6 miRNAs interact with 88 genes, whereas the 9 miRNAs interact with 528 genes. Compared with the genes involved in the Hippo signaling pathway, 4 genes from 88 and 17 from 528 were common (Fig. 2b).

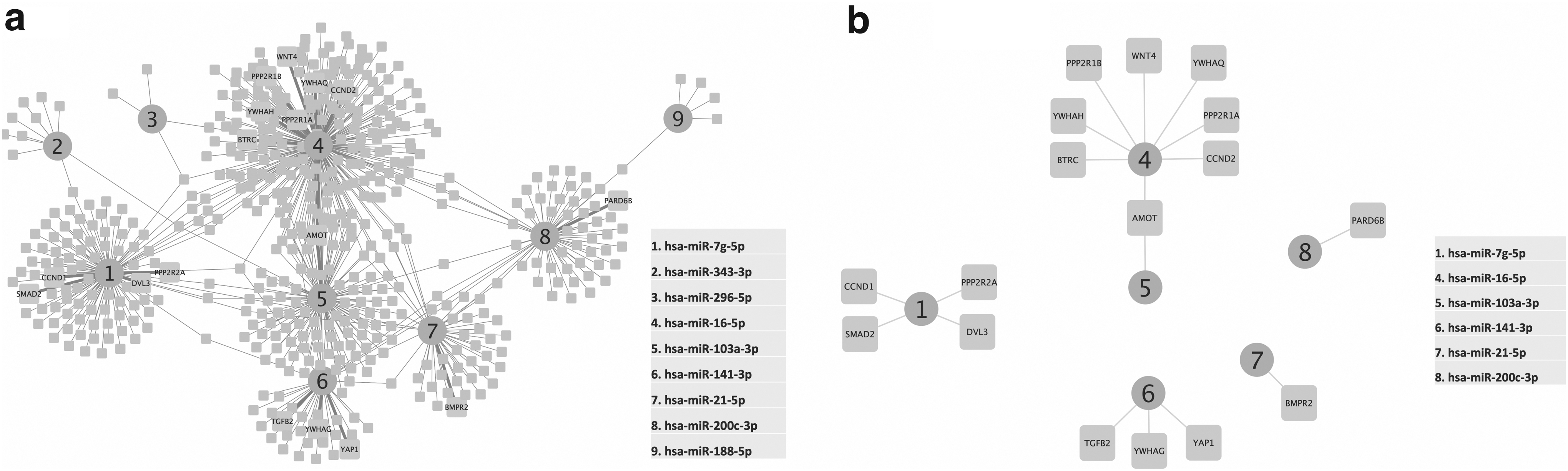
We obtained six upregulated clusters: none had a common interacting gene. hsa-miR-205-5p is the only miRNA interacting with the most genes; four genes are part of the Hippo signaling pathway (Fig. 3). In contrast, nine downregulated miRNAs share common genes and create a network instead of a cluster (Fig. 4a). Six of the nine downregulated miRNAs target 17 Hippo signaling pathway genes. Angiomotin (AMOT) interacts with both hsa-miR-16-5p and hsa-miR-103a-3p (Fig. 4b).

Six upregulated miRNAs with respective targets (Supplementary Table S2). hsa-mir-205-5p (central protein of the cluster, in octagonal shape) is the only miRNA that interacts with the proteins involved in the Hippo pathway (AMOT, PARD68, SMAD1, SMAD4).
Interestingly, when we constructed upregulated miRNA's target's protein–protein interaction network, PARD6B was not part of it. Mothers against decapentaplegic homolog 4 (SMAD4) has the maximum degree of connectivity, whereas AMOT is just coupled to EZR to be part of the network (Fig. 5a–c).

Using the MCL algorithm, we divided the downregulated miRNA's target network into trustworthy clusters and investigated the largest cluster. We found that the PTEN protein has the strongest connection, followed by EP300 and BTRC, a Hippo signaling pathway protein. All three proteins form their hub and are linked with each other with the help of KMT2D and FBXW7 genes (Fig. 5d–f).
GO analysis of the upregulated miRNA-targeting proteins showed that 121 proteins were involved in transcription regulation. Ninety proteins were present in the nucleus, 63 in the cytoplasm and 22 in the membrane, suggesting a distribution of proteins. Further, 128 proteins were involved in the MF of cell division and 87 proteins were involved in protein binding (Supplementary Fig. S1a1–a3 and Supplementary Table S4). GO analysis of the downregulated miRNA-targeting proteins showed that 384 proteins were involved in transcriptional regulation, and 29 were involved in apoptosis.
A total of 416 proteins were present in the nucleus, 430 in the cytoplasm and 101 in the membrane. Further, 512 proteins were involved in protein binding, and 66 and 64 proteins were involved in DNA and RNA binding, respectively (Supplementary Fig. S1b1–b3 and Supplementary Table S5).
KEGG pathway analysis showed that in the case of upregulated miRNAs targeting proteins, EZR and ERBB3 are linked to miRNAs in cancer. YES1 is connected to the adherens junctions. Further, SMAD4 is linked to pancreatic cancer, pluripotency of stem cell-regulating pathways, and adherens junctions (Supplementary Fig. S2a). However, in the case of downregulated miRNAs targeting proteins, BTRC and FBXW7 share Ubiquitin-related proteolysis, BTRC and EP300 share the Wnt signaling pathway, and EP300 and PTEN share five pathways (Supplementary Fig. S2b).
Discussion
In this study, we aimed at finding previously unknown connections within the Hippo signaling pathway and miRNAs to inform the development of novel anticancer treatments. Based on the STRING probability score, the neighborhood principle, and the shortest distance, we found a connection between AMOT and SMAD4, which is illustrated in Figure 5a–c. Both proteins show a role in blood vessel formation, vessel regulation, and cancer control (Huang et al., 2018; Imai et al., 2001; Troyanovsky et al., 2001).
Specifically, SMAD4 is a signal transduction transmembrane protein from the SMAD family. It has an anti-tumor effect by suppressing angiogenesis and increasing blood vessel hyperpermeability. The product of this gene forms homomeric and heteromeric complexes with other activated SMAD proteins, such as SMAD1, that then cluster in the nucleus and regulate the transcription of target genes (Liu et al., 1997; Seong et al., 2007; Yang et al., 2015).
At the same time, AMOT is principally expressed in capillary endothelial cells and larger vessels of the placenta, where it mediates angiostatin's inhibitory influence on tube development and endothelial cell migration toward growth factors during blood vessel formation (Huang et al., 2018; Troyanovsky et al., 2001). According to GO enriched CC analysis, AMOT, EZR, and YES1 are involved in actin formation, which is the basis of new vessel formation (Song et al., 2020; Takeda et al., 2017; Zhang et al., 2019).
Further, in KEGG pathway analysis, SMAD4 is the only link between three separate pathways, namely pancreatic cancer, signaling pathways regulating pluripotency of stem cells, and adherens junction, which confirms the preceding explanation (Supplementary Fig. S2a).
Further, on analyzing the downregulating miRNAs protein–protein network, we found cancer suppression proteins PTEN, EP300, and BTRC (Asaduzzaman et al., 2017; Chen et al., 2018) are forming hubs and are connected directly and indirectly via KMT2D and FBXW7 (Fig. 5d–f). These miRNAs can be included in a cluster of miRNAs being studied to produce new generation miRNA targeting therapies in breast cancer.
According to the KEGG pathway analysis (Supplementary Fig. S2b), PTEN and EP300 share five pathways, three of which are cancer-related; one of which is Hepatitis B, which causes liver cancer; and the last one is the FOXO signaling pathway, which is being explored to identify novel cancer therapies. The Wnt signaling route is shared by EP300 and BTRC; both BTRC and FBXW7 are engaged in the Ubiquitin proteasome pathway, and both are well-studied cancer pathways.
There is an example where increasing the expression of miRNA promotes the expression of tumor-inhibiting proteins. miR-34a is a tumor suppressor miRNA that is often downregulated in breast cancer. It specifically targets Cyclin D1, Bcl-2, and c-Met, all of which are implicated in cancer cell growth. Restoration of miR-34a resulted in increased expression of tumor-inhibiting proteins (Haghi et al., 2019; Rui et al., 2018). In conclusion, increased miRNA expression in breast cancer can promote the expression of tumor-inhibiting proteins by regulating the activity of their target genes.
Conclusions
The dysregulated expression of the Hippo signaling system is characteristic of many cancers, which makes it important in human cancer research. We have uncovered an AMOT-SMAD4 interaction using a multidimensional strategy. However, further research is needed to confirm the interaction. The interaction can be explored for therapeutic targeting to overcome chemoresistance in breast cancer. In addition, we have shown the interconnection of cancer-inhibiting proteins: PTEN, EP300, BTRC, KMT2D, and FBXW7.
The interconnection can be explored further to find a way to upregulate these proteins. In subsequent research, we plan screening and validating compounds that can upregulate the expression of cancer-inhibiting proteins. We suggest that the new age of therapeutics innovation, in part, depends on targeting miRNA. However, targeting a single miRNA is not as successful as targeting a cluster of miRNAs. Our comprehensive bioinformatic analysis provided a list of interacting miRNAs forming clusters. These clusters can also serve as potential biomarkers for early breast cancer diagnosis.
Footnotes
Acknowledgments
The authors thank the principal, Prof. Rajeev Aggarwal, for encouragement and basic infrastructural support at the DBCi4 Centre, Deshbandhu College.
Authors' Contributions
S.R.R.G.: conceptualization, methodology, data curation, visualization, and writing and preparation of original draft. G.N. and P.M.: data curation, figure and first draft preparation. S.R., H.S., and R.S.: data curation, reviewing, and editing. A.S. and I.K.S.: conceptualization, data curation, writing original draft, reviewing, editing, and supervision. All the authors reviewed and made a significant intellectual contribution and approved the final version of the article.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
I.K.S. acknowledges the Indian Council of Medical Research (ICMR) (grant no.33/10/2019-TF/Rare/BMS and ISRM/12(58)/2020 ID no. 2020-1353); S.R.R.G. acknowledges the ICMR; file no.: ISRM/12(58)/2020 ID no. 2020-1353; G.N. and P.M. acknowledge the Council of Scientific and Industrial Research (CSIR), Gov. of India, for providing fellowship.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.