
Abstract
Chronic inflammation is an important contributor to tumorigenesis in many tissues. However, the underlying mechanisms of inflammatory signaling in the tumor microenvironment are not yet fully understood in various cancers. Therefore, this study aimed to uncover the gene expression signatures of inflammation-associated proteins that lead to tumorigenesis, and with an eye to discovery of potential system biomarkers and novel drug candidates in oncology. Gene expression profiles associated with 12 common cancers (e.g., breast invasive carcinoma, colon adenocarcinoma, liver hepatocellular carcinoma, and prostate adenocarcinoma) from The Cancer Genome Atlas were retrieved and mapped to inflammation-related gene sets. Subsequently, the inflammation-associated differentially expressed genes (i-DEGs) were determined. The i-DEGs common in all cancers were proposed as tumor inflammation signatures (TIS) after pan-cancer analysis. A TIS, consisting of 45 proteins, was evaluated as a potential system biomarker based on its prognostic forecasting and secretion profiles in multiple tissues. In addition, i-DEGs for each cancer type were used as queries for drug repurposing. Narciclasine, parthenolide, and homoharringtonine were identified as potential candidates for drug repurposing. Biomarker candidates in relation to inflammation were identified such as KNG1, SPP1, and MIF. Collectively, these findings inform precision diagnostics development to distinguish individual cancer types, and can also pave the way for novel prognostic decision tools and repurposed drugs across multiple cancers. These new findings and hypotheses warrant further research toward precision/personalized medicine in oncology. Pan-cancer analysis of inflammatory mediators can open up new avenues for innovation in cancer diagnostics and therapeutics.
Introduction
Cancer is a complex disease triggered by a complex interplay of genetic and environmental determinants. New ways of thinking about cancer diagnostics and therapeutics are timely and much needed (Turanli et al., 2017; Zhou et al., 2022). In the recent global cancer statistics (GLOBOCAN2020), it has been reported that ∼19.3 million individuals received new cancer diagnoses, and nearly 10.0 million lost their lives because of the disease (Sung et al., 2021). Cancer remains a puzzle in a context of diagnostics and therapeutics innovation, and in part due to efforts to solve the cancer puzzle piece by piece. A systems science approach offers an alternative that can bring in new insights on cancer from basic science to the oncology clinic. In this overarching context, inflammation is one of the cancer pathogenesis-related dimensions wherein a systems approach might be particularly fruitful. Of note, the relationship between inflammation and the promotion of cancer was first observed in the 19th century by Virchow, who indicated that cancer may result from chronic inflammation (Balkwill and Mantovani, 2001), but only in recent years has this become a widely recognized phenomenon (Nigam et al., 2023).
Inflammation is a natural reaction of the body to both internal and external factors in the environment such as physical tissue damage, infection, and other challenges to the immune system. The inflammatory process eliminates and regulates tissue physiology. When homeostasis is disrupted, macrophages and mast cells release bioactive stimuli such as cytokines, chemokines, reactive oxygen species (ROS), and histamine. These conditions at the site of injury associated with the inflammatory process lead to mobilization of leukocytes and leakage of blood (De Visser et al., 2006). However, the inflammatory process is a double-edged sword. When these reactions persist over a long period of time, it leads to chronic inflammation, which results not only in some selected diseases such as coronary heart disease, diabetes, intestinal disease, and arthritis but also in a wide range of mental and physical disorders that prevail in the current global morbidity and mortality statistics (Du et al., 2015; Furman et al., 2019; Medzhitov, 2010; Sung et al., 2021). Many clinical and epidemiological studies have also demonstrated a strong association between inflammation and cancer (Du et al., 2015; Munn, 2017; Zhao et al., 2021).
Similar to cancer, chronic inflammatory diseases are also recognized as one of the leading causes of death, as more than half of all deaths are due to inflammation-related diseases (Furman et al., 2019; Roth et al., 2018).
Given the linkages between tumor formation and the attendant crucial role of inflammation (Dobrovolskaia and Kozlov, 2005; Nigam et al., 2023), unpacking the molecular mechanisms of inflammation in cancers would be a promising avenue for cancer diagnostics and therapeutics innovation. In some cancers such as gastric cancer and mucosa-associated lymphoma caused by viral influences, the inflammatory response triggered by infection might prevent tumor development and serves initially as a defense mechanism aimed at eliminating the pathogen in acute inflammation (Karin, 2006; Wu et al., 2009). On the other hand, immune dysfunction and autoimmunity contribute to chronic inflammation that drives tumor growth. A notable example of this association can be observed in inflammatory bowel disease, which has been shown to substantially elevate the susceptibility to colorectal cancer. These prior observations underscore the intricate interplay between immune dysfunction, chronic inflammation, and the promotion of tumorigenesis (Waldner and Neurath, 2009).
Chronic inflammation, often linked to infection or autoimmune disorders, plays a pivotal role in the initiation of tumorigenesis by promoting oncogenic mutations, genetic instability, early stages of tumor promotion, and augmented angiogenesis. In essence, the inflammatory response contributes to the facilitation of neoangiogenesis, tumor advancement, and metastatic dissemination, while also fostering genomic instability through localized immunosuppression (Zhao et al., 2021). The specificity of immune cells is related to the spread of cytokines and chemokines against external agents in the tumor microenvironment (Hibino et al., 2021). For example, effectors such as NF-kB, AP-1, STAT, and SMAD transcription factors as well as caspases and cytokines control immunity and antitumor activations. IL-6, IL-17, and IL-23 cytokines play roles in tumor progression. TRAIL, FasL, TNF-a, EGFR ligands, TGF-b, and IL-6 increase cancer cell formation and survival (Lin and Karin, 2007). Other mechanisms related to inflammation and cancer are inflammation-induced mutagenesis and ROS.
These two mechanisms can inactivate or suppress repair enzymes or they can inactivate key tumor suppressor genes, such as Tgfbr2 and Bax, to protect cancer cells (Colotta et al., 2009; Hussain et al., 2003).
As noted earlier, systems science is an important accelerator in cancer-related discoveries and translational research (Gulfidan et al., 2022; Gulfidan et al., 2021; Turanli et al., 2019a; Turanli et al., 2019c; Ulgen et al., 2023). Recent pan-cancer studies under the systems biology perspective not only reveal novel protein–protein interactions (PPIs) to distinguish cancer types but also decipher common PPI modules that trigger tumor formation (Gulfidan et al., 2020; Kori et al., 2022a; Kori et al., 2022b). In the current era of big data and data deluge, there are both opportunities and challenges in moving discoveries at the intersection of cancer research and systemic or chronic inflammation (Kori et al., 2022b).
The aim of the present study was to uncover the gene expression signatures of inflammation-associated proteins that lead to tumorigenesis, and with an eye to discovery of potential system biomarkers and novel drug candidates in oncology. For that purpose, inflammation-associated differentially expressed genes (i-DEGs) are determined in this study and common i-DEGs are proposed as tumor inflammation signatures (TIS) after pan-cancer analysis. After examining DEGs, i-DEGs are determined for each cancer type and used for drug repurposing. The prognostic features and secretion profiles of TIS were determined to follow cancer progression invasively. Eventually, this study offers candidate inflammation-associated biomarkers that may be useful for differentiating cancer types or domineering in multiple cancers to develop novel prognostic tools and repurposed drugs for ameliorated cancer therapy based on inflammation signatures.
Materials and Methods
Pan-cancer gene expression data collection
In this study, transcriptome data of 12 different cancers comprising breast invasive carcinoma (BRCA), colon adenocarcinoma (COAD), head and neck squamous cell carcinoma (HNSC), kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), prostate adenocarcinoma (PRAD), stomach adenocarcinoma (STAD), thyroid carcinoma (THCA), and uterine corpus endometrial carcinoma (UCEC) were extracted from the Cancer Genome Atlas (TCGA) (Tomczak et al., 2015). Data were collected due to the sample number criteria in both tumor and control groups (n > 30) (Gulfidan et al., 2020; Kori et al., 2022a; Oktem et al., 2023). The count data were collected by using TCGABiolinks (Colaprico et al., 2016). In total, 6876 gene expression profiles were collected from 6239 primary tumors and 637 matched normal tissue samples. All cancer types in this study and their sample numbers are represented in Figure 1A.
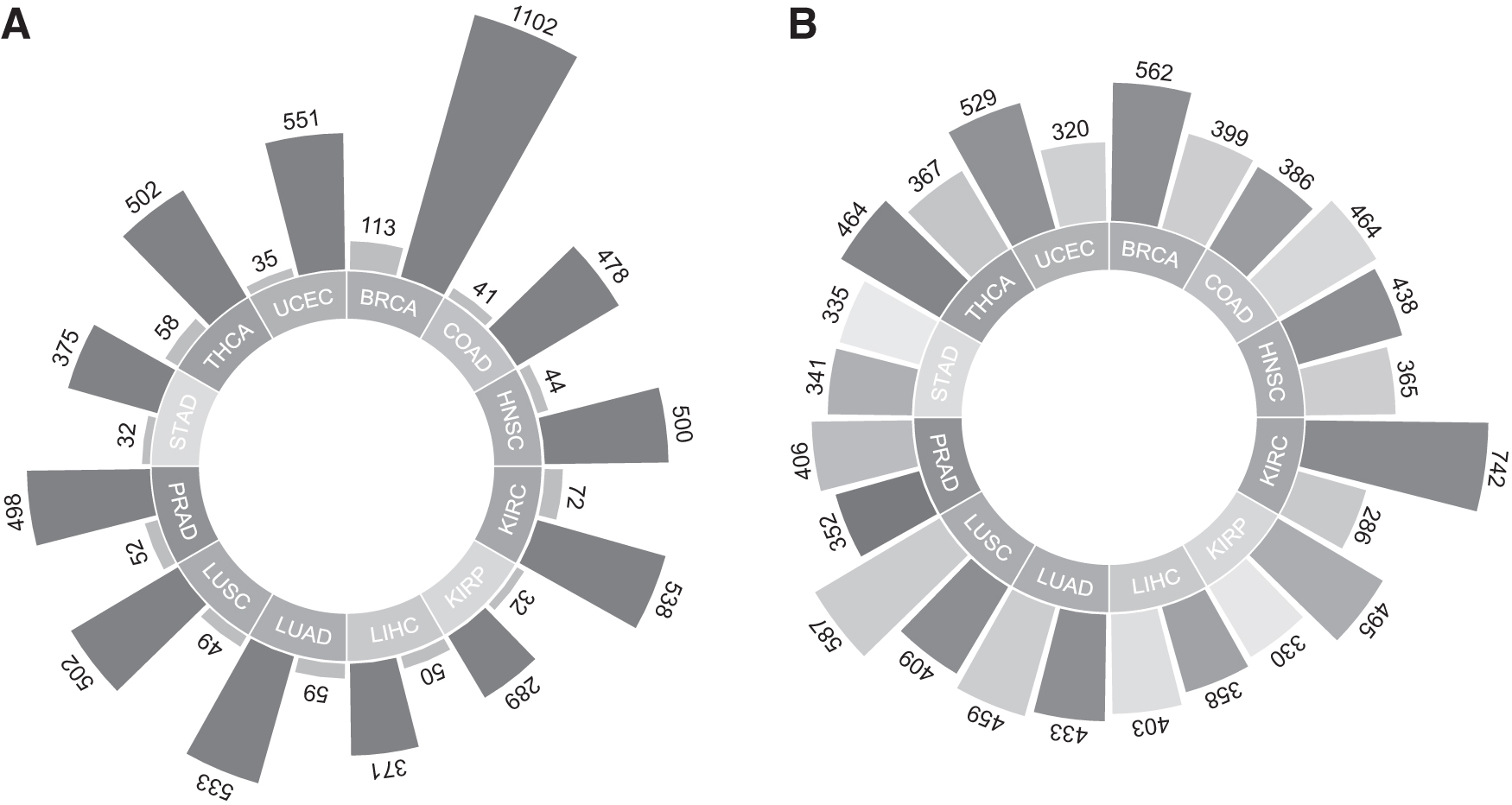
Pan-Cancer data collection and results of differential expression analysis
Determination of inflammation-associated proteins
A compilation of inflammation-associated proteins was assembled following a methodology similarly to our previous study (Kori et al., 2022a). Initially, proteins categorized under the inflammatory response process (GO: 0006954) were identified by employing QuickGO, a web-based tool that facilitates the search for Gene Ontology (GO) annotations (version 2021-01-08) (Binns et al., 2009). Furthermore, the UniProt portal (Coudert et al., 2023) was utilized to search for proteins associated with inflammation using the keyword “inflammation.” As a result, a comprehensive list of 1298 inflammation-associated proteins was generated (Supplementary Table S1).
Identification of DEGs
Differential expression analysis was performed using the R/Bioconductor package “DESeq2” manual (Love et al., 2014) to identify genes with significant expression differences between two phenotypes, utilizing raw count data as performed in the recent study (Oktem et al., 2023). Multiple hypothesis correction used in this study was Benjamini and Hochberg false discovery rate. Genes with the FDR-adjusted p-value <0.05 were considered as DEGs. Positive logFC ratios (logFC >0) were used to determine upregulated genes and negative logFC ratios (logFC <0) were used for downregulated genes (Supplementary Table S2) similarly to our previous studies (Gulfidan et al., 2022; Turanli et al., 2019b). The DEGs from the 12 cancer types were combined with a curated list of genes encoding proteins associated with inflammation. Through this integration, i-DEGs were identified (Fig. 1B). The subset of i-DEGs shared across all cancer types was referred to as TISs.
Functional enrichment analysis and protein function
The pathway and functional enrichment analyses were carried out through Metascape (Zhou et al., 2019) to identify functional annotations significantly associated with the DEGs. The KEGG Pathway (Kanehisa et al., 2017), GO (The Gene Ontology Consortium, 2019), and Reactome (Fabregat et al., 2018) databases were used as the data source of biological processes and molecular functions. Additionally, the druggability and functions of TISs were screened using the Drug Gene Interaction Database (DGIdb v4.2.0) (Cotto et al., 2018). All enriched GO terms and pathways were listed in Supplementary Table S3.
Prognostic performance analyses of TISs
To assess the prognostic value of TISs in different cancer types, survival analyses were conducted based on clinical information using RNA-Seq data obtained from TCGA datasets, following similar methodologies as previous studies (Gulfidan et al., 2020; Kori et al., 2022b). The survival package in R (version 4.0.2) was used to present the survival analyses. The study participants were categorized into low- and high-risk groups based on their prognostic index, also known as the risk score. Kaplan–Meier plots were employed to illustrate the survival patterns associated with the identified signatures in each group. A log-rank p-value threshold of <0.05 was considered statistically significant for determining the cutoff point.
Secretion profiles of TISs
Since TISs are proposed as systems biomarker candidates to screen by either invasive or noninvasive techniques, the level of the proteins coded by TIS was investigated. The secretion levels (ppm) of TISs in serum, plasma, peripheral blood mononuclear cell, adipocyte, urine, saliva, hair follicle, oral epithelium, and nasal respiratory epithelium were explored through protein expression data, which are accessible in the GeneCards (Safran et al., 2021) database curating the proteomics databases comprising ProteomicsDB (Samaras et al., 2019), MaxQB (Schaab et al., 2012), and MOPED (Kolker et al., 2013).
Drug repurposing based on TISs
To explore the drugs that may have effects on the reversal of gene signatures in disease, the L1000CDS2 (Duan et al., 2016) search engine was employed for 12 cancer types individually by using i-DEGs as a query. Considering the direction of gene expression patterns (up- and downregulated genes), candidate drugs were found and ranked by their scores. The top 50 drugs for each cancer type were selected for further evaluation. The repurposed drugs were further investigated through the literature search and publicly available drug databases such as DrugBank (Wishart et al., 2018).
Results
Pan-cancer analysis of inflammation-associated DEGs
First, the inflammation-associated protein list (Supplementary Table S1) was constructed as similarly to our previous study (Kori et al., 2022a) to identify inflammation signatures. The current number of proteins that are associated with inflammation was determined as 1298. Second, DEGs were determined for 12 human cancers, separately. Later, the DEGs of 12 cancer types were mapped to the generated list of genes coding inflammation-associated proteins, and i-DEGs were identified and presented (Supplementary Table S2).
Numbers of the i-DEGs for each cancer type are shown in Figure 1B. The highest number of DEGs (1028) was in KIRC comprising 742 upregulated and 286 downregulated; the lowest number of the DEGs (676) was in STAD comprising 335 downregulated and 341 upregulated in this study (Fig. 1B). According to the total i-DEGs' number, LUSC (996), BRCA (961), LUAD (892), and COAD (850) follow KIRC. The common i-DEGs in 12 human cancers were 45 and denoted as TIS. The logFC of TIS gene expressions was represented as a heatmap (Fig. 2A) and 65% of the genes were upregulated, whereas 35% of them were downregulated in all cancers. According to the direction of TIS expression, KIRC and KIRP, LUAD, and LUSC were clustered while their origin organs were also the same. Surprisingly, PRAD and STAD; COAD and HNSC; and BRCA and LIHC were clustered first (Fig. 2A). Enriched GO terms/KEGG pathways are shown by bar plot representation (Fig. 2B). Not surprisingly, regulation of inflammatory response, regulation of leukocyte activation, and inflammatory response were at the top of enriched GO terms.
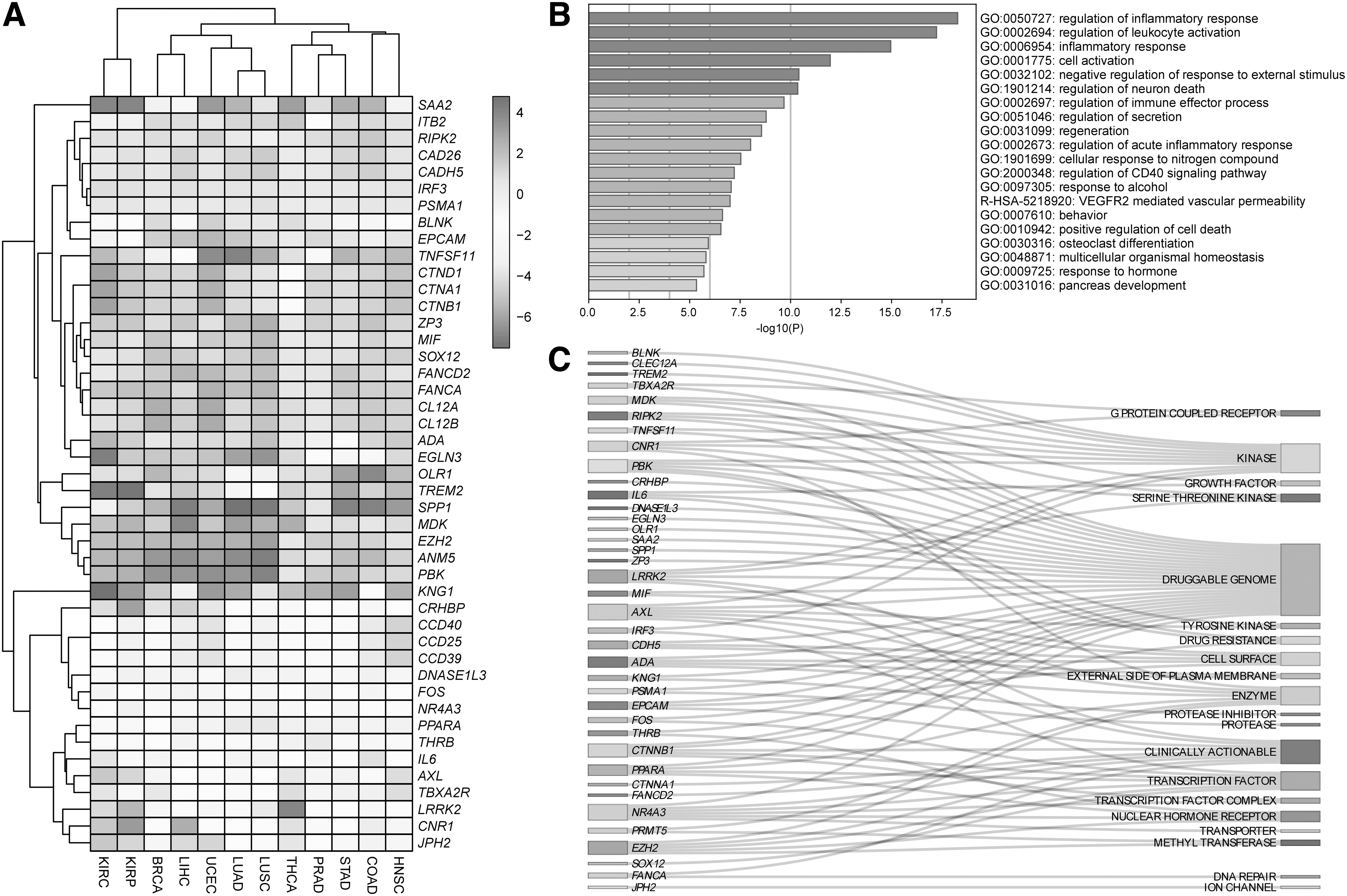
Common i-DEGs in 12 human cancers denoted as TIS in the current study
Besides, VEGFR2-mediated vascular permeability, response to alcohol, regulation of neural death, and positive regulation of cell death were other enriched terms and pathways regarding TISs. All other details on functional enrichment analysis are listed in Supplementary Table S3.
In addition to focusing on common i-DEGs, the distinct inflammatory signatures may also lead to propose targeted therapy options or early diagnosis for various cancer types. The gene expression profiles of 1298 inflammation-associated protein-coding genes are listed in Supplementary Table S2. There are only two sets of i-DEGs that were found significant in only one cancer type. A group of genes consisting MRGPRX1, LCE3C, PYDC2, and PYDC5 were determined as differentially expressed only in LUSC. Another set of genes consisting DRB1, DRB4, DRB5, and GPR32 were determined as differentially expressed only in KIRC.
Tumor inflammation signatures were also searched through DGIdb to reveal druggable genes. Sixty percent of TIS was denoted as druggable genome. Eleven of 45 proteins (AXL, BLNK, CLEC12A, CNR1, IRF3, LRRK2, MDK, PBK, RIPK2, TNFSF11, TREM2) were kinases; 9 of them (AXL, CTNNA1, CTNNB1, EPCAM, EZH2, FANCA, FANCD2, LRRK2, NR4A3) were clinically actionable; 7 of 45 (ADA, PRMT5, AXL, CNR1, EZH2, LRRK2, PPARA) were also enzyme, and 7 of them (CTNNB1, EZH2, FOS, IRF3, NR4A3, PPARA, SOX12) were transcription factors (Fig. 2C). All results on the functional grouping of TIS proteins are presented in Supplementary Table S4.
Interpreting prognostic performance of TISs and their secretion profiles
In the current study, TISs comprising 45 i-DEGs were examined for their possibility as systems biomarkers. Although there is strong evidence linking inflammation to various cancers (Hou et al., 2021; Mei et al., 2014; Yu et al., 2022), there are no robust biomarkers to track cancer progression. However, inflammation may also influence the prognosis of various cancers. Therefore, it was hypothesized that TISs can be used as system biomarkers and show high prognostic performance in cancer patients. According to the survival analyses, TIS had significantly high prognostic power in 12 human cancers analyzed (Fig. 3). The lowest p-value was in KIRC while the highest hazard ratio was 13.34 in THCA.
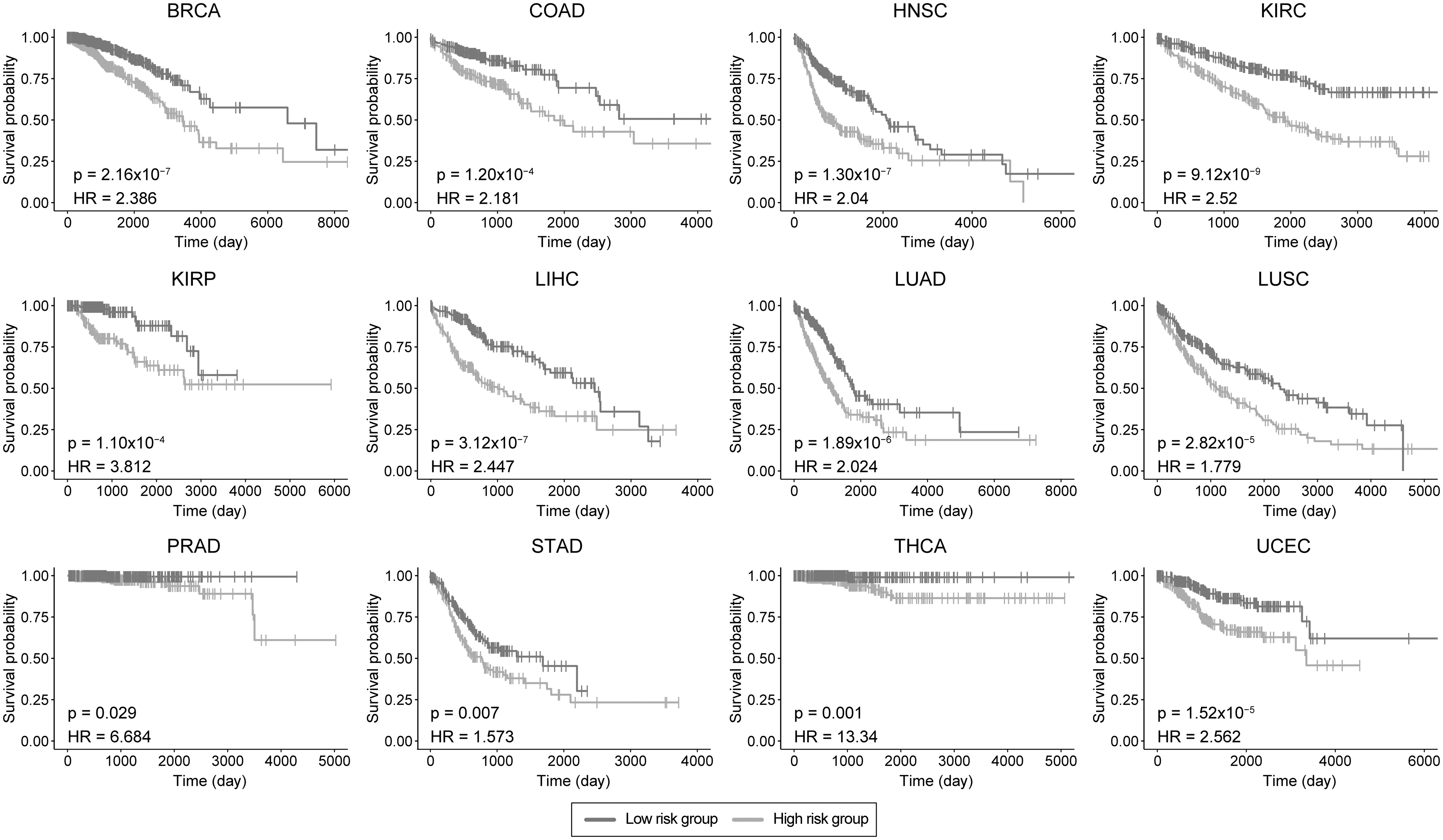
Prognostic power of TISs for each cancer type. Kaplan–Meier Plots estimate patients' survival for 12 cancers with p-value and hazard ratio given for each curve.
Another important issue is biomarker monitoring in clinical care. It is more favorable to use noninvasive methods than invasive ones. If not, there are more favorable tissues such as blood for invasive methods to track biomarker levels. Secretion profiles from TIS were sought in tissues that are favorable and relatively easy to collect. Thirteen of 45 TIS (SPP1, SAA2, PSMA1, MIF, KNG1, ITB2, EPCAM, CTND1, CTNB1, CRHBP, CADH5, AXL, ADA) were secreted only in different tissues such as serum, plasma, urine, saliva, etc. AXL, CADH5, CRHBP, SPP1, SAA2, PSMA1, and KNG1 were secreted into serum. The concentration of KNG1, encoding inhibitors of thiol proteases, was about 5000 ppm in serum, plasma, and urine. The highest protein concentration detected belonged to SPP1 protein in the urine. The secretion profiles of the proteins encoding TIS are shown as a bubble diagram in Figure 4. All results on secretion profiles of TIS proteins in ppm level are presented in the supplementary file as well (Supplementary Table S4).
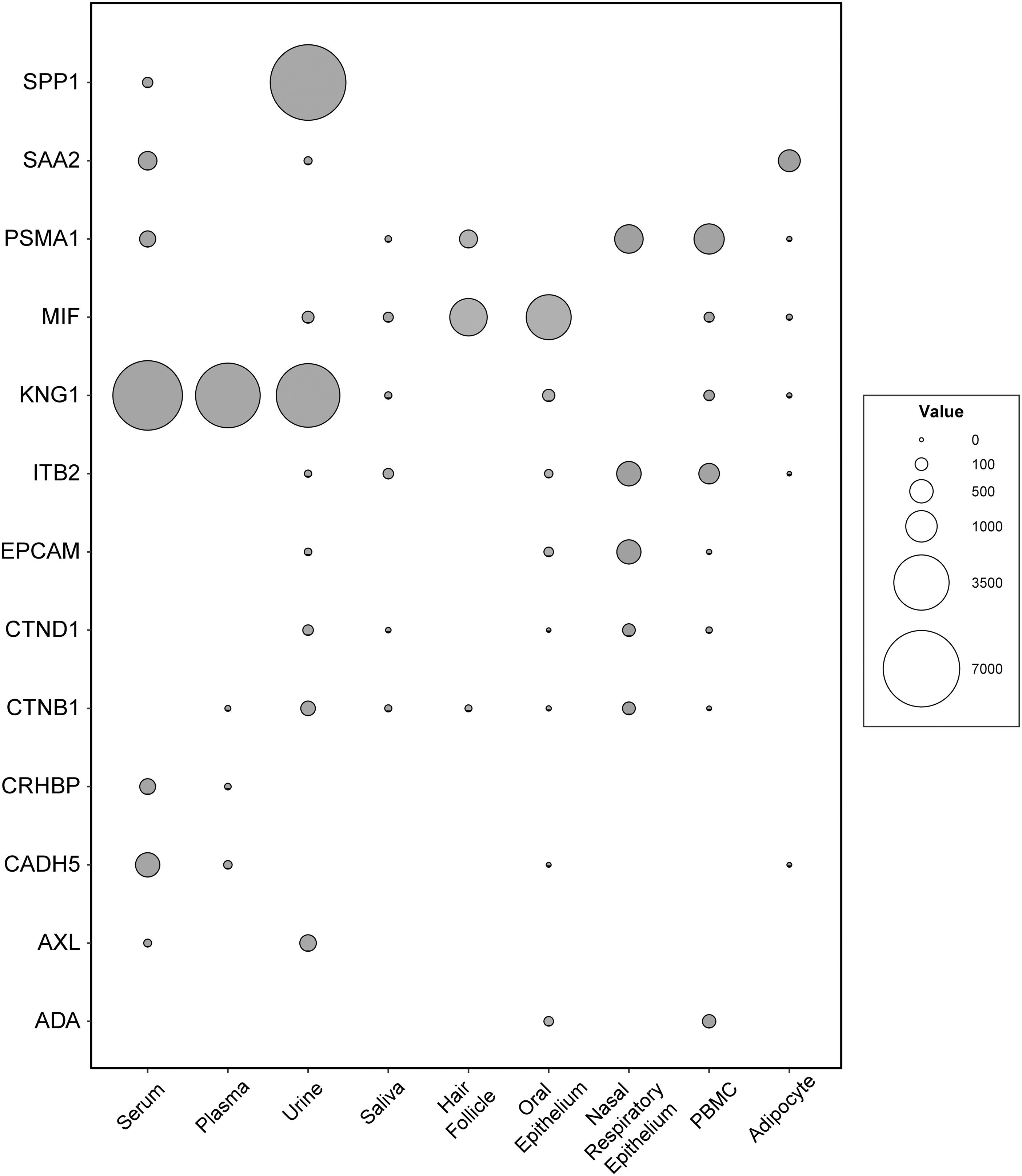
Secretion profiles of TIS. The detection concentrations (ppm) for protein coding TIS were presented as a bubble plot. X axis shows the tissues that is relatively easier to collect samples, whereas Y axis shows the proteins that are secreted.
Repurposed drugs based on TISs
L1000CDS2 is used to identify small molecules that can either reverse or mimic gene expression based on the given input of differently expressed genes (Duan et al., 2016). To reveal the drug candidates reversing the gene signatures of each cancer type, i-DEGs were used as input queries. The top small molecules identified by the L1000CDS2 query are listed in detail for each cancer type, separately (Supplementary Table S5). Two hundred thirty-eight unique small molecules were repurposed in 12 human cancer types, totally. Thirteen of 238 small molecules comprising narciclasine, selumetinib, BRD-K92317137, emetine hydrochloride, homoharringtonine, V4877, withaferin-a, BRD-K28907958, BRD-K76674262, emetine dihydrochloride hydrate, etoposide, LDN-193189, and parthenolide were determined in at least five different cancer types and represented in Figure 5. Narciclasine, a plant growth inhibitor isolated from bulbs of several Narcissus species (Lv et al., 2022) was the most common molecule in nine different cancer types, including BRCA, KIRP, LIHC, LUAD, LUSC, PRAD, STAD, THCA, and UCEC.
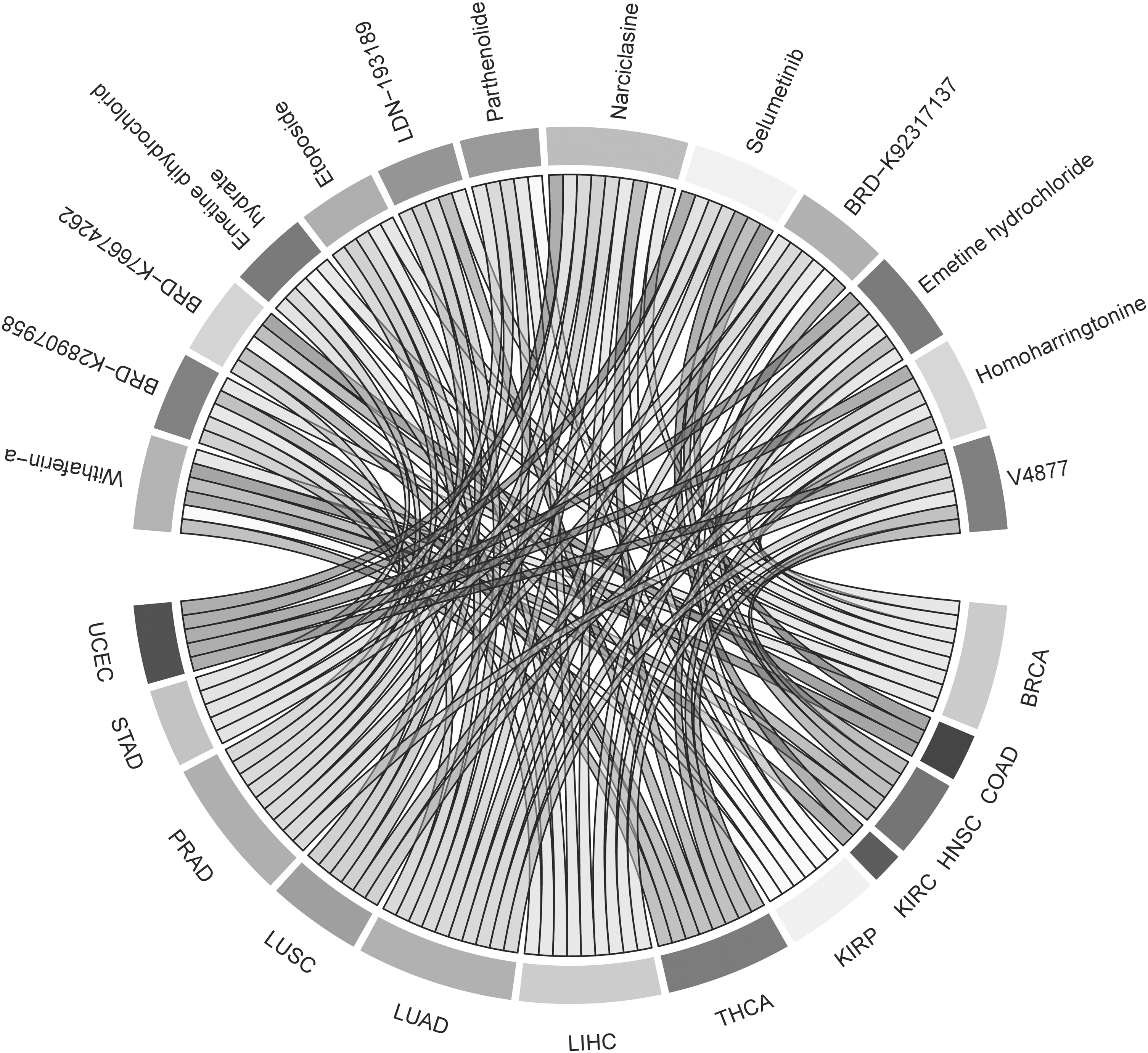
Repurposed small molecules determined at least five cancer types due to the results acquired from L1000CDS2 tool.
Discussion
Cancer is a common complex disease with genetic and environmental underpinnings. Pan-cancer studies have been reported in the literature that aim to uncover the molecular substrates of cancers. These studies also inform the person-to-person and between-population differences in cancer pathogenesis, and by extension new ways to develop individually tailored precision/personalized medicines. Precision/personalized medicine encompass a broad range of activities to advance the disease diagnosis, prognosis and therapeutics, including in oncology (Campbell et al., 2020; Gulfidan et al., 2020; Kori et al., 2022b; Li et al., 2022; Liu et al., 2022a; Wen et al., 2021).
Seen through the lens of cancer precision/personalized medicine, computational and systems biology studies of inflammation is timely to better understand the tumor microenvironments that lead to different clinical and therapeutic outcomes. Acute inflammation usually prevents cancer development, whereas chronic inflammation promotes carcinogenesis. Although some cancers are not directly related to inflammation, inflammatory responses are known to influence every aspect of cancer development and progression, including DNA replication, tumor cell survival, angiogenesis, metastasis, and undermining adaptive immunity (Philip et al., 2004).
The present study included 12 human cancers while the previous studies on the relationship between inflammation and particular cancer types were also searched. The highest DEG number was shown in KIRC, which is already reported as an inflammation-related carcinoma. In KIRC, inflammation has been recognized as a crucial component in inducing tumorigenesis. In another study based on gene expressions, the role of inflammation in KIRC was verified through inflammation-associated gene correlations with the tumor microenvironment (Yu et al., 2022). The importance of an inflammatory microenvironment for carcinogenesis was studied through the activation profiles of immune cells and stromal cells in CRC (Schmitt and Grete, 2021). Although the role of chronic inflammation in the initiation and development of breast cancer is unclear, there is a strong correlation between BRCA clinical outcomes as the risk factor for the transition to malignancy (Danforth, 2021). In prostate cancer, inflammation increased the risk of transition of benign prostate biopsy specimens to high-grade prostate tumors in adjacent areas (Stark et al., 2015).
The inflammatory tumor microenvironment around the lung plays an important role in tumorigenesis, which become the most common and fatal malignant tumor in the world (Tan et al., 2021). Chronic inflammation is also developed by persistent infections in the body. For example, Helicobacter pylori infection is the primary cause of gastric cancer and contributes to carcinogenesis in the stomach (Piazuelo et al., 2019). Therefore, it is not surprising to obtain inflammatory gene signatures that are differentially expressed in cancer cases. How to control inflammation before promoting carcinogenesis is the nitty gritty of further scientific efforts.
Although several studies have investigated the essential role of inflammation in tumor progression (Munn, 2017; Murata, 2018; Philip et al., 2004), systematic pan-cancer analysis of gene expression patterns related to inflammation is limited.
In a study linking inflammation and various cancers at the gene expression level, three groups of cancer were identified according to differential gene expression patterns on inflammation signatures covering early onset, transition, and late phase of inflammation phenotypes. Their results under the context of inflammation provide an influential background for identifying differences in gene expression in various cancers (Yu et al., 2014). In another study, gene signatures comprising 18 genes were defined as TISs that evaluate a pre-existing but repressed adaptive immune response in tumors. The expression patterns of the genes were conserved across tumor types but appeared to have a low prognosis in most cancers, so they may serve as a pan-cancer measure of the inflamed tumor phenotype. These inflammatory signatures could lead to superior indication selection for suitable immunotherapeutics (Danaher et al., 2018).
Tumor inflammation signatures comprising 45 inflammation-associated DEGs were proposed as biomarker candidates that serve high prognostic performance on each cancer type in this study. Several of our findings are also confirmed by previous studies. SPP1, also called osteopontin, is an extracellular secreted glycol phosphoprotein. Recent studies reported the abnormal expression of osteopontin in various malignancies, which is closely associated with the proliferation, migration, and invasion of tumors (Yi et al., 2022). Another protein proposed in this study, ANM5, is a protein methyl transferase translated by PRMT5 gene. Typically, protein methyl transferases play essential roles in controlling pathways associated with the development, advancement, and response to therapy in cancer. The impact of PRMT5 on histone methylation and methylation of regulatory proteins forms the basis for cancer development through metabolic signaling, RNA splicing, cell cycle regulation, and apoptosis.
Due to these factors, the significance of regulating PRMT5 and focusing on it as a target for cancer treatment were emphasized (Kim and Ronai, 2020). A vital gene involved in the control of mitosis and tumorigenesis in different types of cancer is PDZ-binding kinase (PBK), a serine/threonine kinase belonging to the mitogen-activated protein kinase kinase family.
The prognostic and predictive value of PBK expression in 33 cancer types were systematically analyzed in a previous study (Wen et al., 2021). Enhancer of zeste homolog 2 (EZH2) is the enzymatic catalytic subunit of polycomb repressive complex 2 (PRC2) that functions in cell proliferation, apoptosis, and cell senescence. Therefore, targeting EZH2 for cancer therapy and discovering different types of EZH2 inhibitors have taken attention. The role of EZH2 in cancer initiation, progression, metastasis, metabolism, drug resistance, and immunity regulation were reviewed, recently (Duan et al., 2020). Midkine (MDK) is a growth factor with an affinity for heparin, and its expression is dysregulated in several human malignancies. It serves as a crucial mediator in acquiring essential characteristics of cancer, such as enhanced cell proliferation, prolonged survival, increased metastatic potential, augmented cellular migration, and promotion of angiogenesis (Filippou et al., 2020).
Subsequently, the secretion profiles of these genes were evaluated for a clear-cut transition to the clinic. Not all proteins were found in serum, plasma, and other tissues. However, the SPP1 gene has been found as overexpressed in cancer types, such as BRCA, LIHC, and LUAD, similarly to this study. In cancer progression, the role of SPP1 in tumor cell growth, migration, and invasiveness, as well as chemoresistance has been reported (Liu et al., 2022b). KNG1 protein was proposed as a serum protein biomarker of colorectal adenoma in patients with a family history of colorectal cancer (Yu et al., 2018). The Macrophage Migration Inhibitory Factor (MIF) is an inflammatory cytokine that is overexpressed in several cancer types in parallel with this study. Increased expression of MIF correlated with tumor aggressiveness and poor patient outcomes (Balogh et al., 2018).
Drug repurposing is also performed regarding the i-DEGs from each cancer type. Top repurposed drugs highlighted plant alkaloids that act specifically by blocking the ability of a cancer cell division (Efferth and Oesch, 2021). Narciclasine is at the top of the list and targeted to nine cancer types, except for COAD, HNSC, and KIRC. Narciclasine is a natural product found in Amaryllidaceae family and belongs to a class of molecules named “isocarbostyril alkaloid.” The drug displays strong cytotoxicity effects against a variety of cancer cells such as non-Hodgkin lymphoma subtype, and primary effusion lymphoma (Gopalakrishnan et al., 2020). One of the recent studies showed that STAT3 was the direct target of narciclasine (Lv et al., 2022). This study also supported this finding based on narciclasine repurposed for the cancer types where STAT3 was differentially expressed.
Homoharringtonine is another plant alkaloid that has been repurposed in this study. It has also antitumor properties widely used for the treatment of hematologic malignancies such as chronic myeloid leukemia, and acute myeloid leukemia (Lü and Wang, 2014). The compound Parthenolide (PN) has been uncovered to hinder NF-κB signaling and additional survival-promoting pathways. It also decreases cancer stem-like cells by stimulating apoptosis in various types of cancer. Utilizing PN or its derivatives in combined therapeutic strategies seems to hold potential as a method to heighten responsiveness to cancer treatment and diminish resistance. (Sztiller-Sikorska and Czyz, 2020). PN with promising anticancer activity is a major component derived from feverfew (Tanacetum parthenium). It has been shown to promote apoptotic cell death in various cancer cells (Liu et al., 2017).
The cancer drug repurposing studies were not limited to revealing reversal gene expressions, and there are many different computational methods applied such as causal inference–probabilistic matrix factorization (Yang et al., 2014) or machine/deep learning approaches such as deepDR (Zeng et al., 2019). The different approaches and tools used to repurpose drugs for cancer therapy were already detailed (Turanli et al., 2019). It is noteworthy, as a limitation of the present study, the lack of experimental validations of the findings and the hypotheses generated, which call for further translational research in the future. However, the findings and i-DEGs reported herein in particular, inform future in vitro and translational clinical studies toward cancer precision medicine (Supplementary Table S2).
Conclusions
In this study, differential expression patterns of inflammation-associated signatures in 12 human cancer types were retrieved from TCGA and common signatures were evaluated across their secretion profiles and prognostic powers. In addition, drug repurposing was carried out based on inflammatory gene expression patterns for each cancer type. However, in vitro and in vivo studies, including large-scale clinical research, are required so as to translate the present computational findings to the clinic and public health practice. While cancer biomarker research is a very active field of scientific inquiry, there are only limited number of pan-cancer analyses of inflammatory signatures despite the significant role inflammation plays in cancer pathogenesis. The present study suggests that the generalized TISs are promising as potential prognostic markers and their secretion profiles in the 12 human cancer types studied in this research.
While the overarching goal of precision medicine is to formulate personalized medical interventions, it remains imperative to establish standardized biomarkers that can exhibit individual variability. These parameters serve as foundational reference points, accommodating the inherent diversities inherent in each patient's unique genetic makeup, molecular profiles, environmental exposures, and lifestyle factors.
In all, this study generates new findings and hypotheses that warrant further research toward precision/personalized medicine in oncology. The present study shows TISs as potential biomarkers that can be used for early detection of cancer or a higher risk of developing cancer, allowing for timely interventions and better outcomes. Moreover, understanding these biomarkers can lead to the development of targeted therapies that specifically address the inflammatory processes driving cancer growth. Biomarkers can also be used to monitor treatment progress and predict the likelihood of disease recurrence. Therefore, the findings underscore pan-cancer analysis of inflammatory mediators as an important avenue for innovation in cancer diagnostics and therapeutics.
Footnotes
Acknowledgment
The author hereby expresses gratitude to Gizem Gulfidan for her unwavering support and invaluable discussions.
Authors' Contributions
B.T. envisaged the study, executed the analyses, interpreted the results, and wrote the article.
Author Disclosure Statement
The author declares there are no conflicting financial interests.
Funding Information
No funding was received for this article.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.