
Abstract
Klebsiella pneumoniae is an opportunistic multidrug-resistant bacterial pathogen responsible for various health care-associated infections. The prediction of proteins that are essential for the survival of bacterial pathogens can greatly facilitate the drug development and discovery pipeline toward target identification. To this end, the present study reports a comprehensive computational approach integrating bioinformatics and systems biology-based methods to identify essential proteins of K. pneumoniae involved in vital processes. From the proteome of this pathogen, we predicted a total of 854 essential proteins based on sequence, protein–protein interaction (PPI) and genome-scale metabolic model methods. These predicted essential proteins are involved in vital processes for cellular regulation such as translation, metabolism, and biosynthesis of essential factors, among others. Cluster analysis of the PPI network revealed the highly connected modules involved in the basic functionality of the organism. Further, the predicted consensus set of essential proteins of K. pneumoniae was evaluated by comparing them with existing resources (NetGenes and PATHOgenex) and literature. The findings of this study offer guidance toward understanding cell functionality, thereby facilitating the understanding of pathogen systems and providing a way forward to shortlist potential therapeutic candidates for developing novel antimicrobial agents against K. pneumoniae. In addition, the research strategy presented herein is a fusion of sequence and systems biology-based approaches that offers prospects as a model to predict essential proteins for other pathogens.
Introduction
Klebsiella pneumoniae is an opportunistic bacterial pathogen that causes several hospital-acquired infections, including pneumonia, urinary tract infection, bacteremia, and so on, in immunocompromised patients in health care settings (CDC.gov, 2023; Chang et al., 2021). In recent years, this pathogen has become a significant public health threat due to the emergence of multidrug-resistant strains and hypervirulent strains, which have been reported in various parts of the world (Awoke et al., 2021; Banerjee et al., 2021; Chen et al., 2023). Indeed, K. pneumoniae has developed resistance to multiple antibiotics, including the last resort regimens such as colistin and carbapenems, with various resistance mechanisms (Petrosillo et al., 2019). The increased mortality associated with multidrug-resistant strains of K. pneumoniae demands the development of therapeutic intervention in treating the infections caused by this pathogen (Li et al., 2023; Xu et al., 2017).
In this scenario, several studies attempted to identify potential targets and drugs in K. pneumoniae using various omics-based approaches (Ali et al., 2022; Ramos et al., 2018; Serral et al., 2022) and other studies to enhance existing treatment options (Bayatinejad et al., 2023; Shein et al., 2021).
For effective therapeutics discovery, exploration of essential genes/proteins of a pathogenic organism can assist us in comprehending the pathogen system and pinpoint key factors that are vital to various key biological processes (Plaimas et al., 2010). The proteins encoded by the essential genes in an organism are termed as essential proteins that are indispensable for the growth and survival of the organism. The identification of such essential proteins in a pathogen would facilitate in shortlisting potential target candidates, which can be further utilized for developing/designing antimicrobial agents. Multiple subtractive genomics-based studies on pathogenic bacteria use this strategy to filter potential candidate targets for drug target identification and vaccine development (Khan et al., 2022; Shanmugham and Pan, 2013; Solanki and Tiwari, 2018; Uddin et al., 2018).
These studies mainly relied on the sequence-based approach that compares the pathogen proteins with a known set of essential proteins to select proteins for further analysis. Similarly, there are reports on network-based methods and genome-scale metabolic modeling (GSSM) approaches for the identification of essential genes/proteins in various organisms (Gollapalli et al., 2021; Sertbas and Ulgen, 2020; Wuchty and Uetz, 2014).
Researchers have developed and applied several approaches encompassing both in vitro and in silico analysis to identify essential genes/proteins in various organisms, ranging from archaea, bacteria to eukaryotes (Guo et al., 2021). The identification methods have evolved to utilize various omics data and use machine/deep learning-based approaches, which necessitate high-quality large datasets for such prediction (Aromolaran et al., 2021; Yue et al., 2022). In this scenario, with the availability of multiple biological data, including proteome sequence, interactome, and so on, an array of combination analyses would be advantageous as the predictions are consolidated results from different methods. Moreover, prediction of essential proteins in K. pneumoniae utilizing a fusion of sequence and systems biology-based integrative approach is absent.
To this end, the present study aims to predict essential proteins in this pathogen using an integrative computational approach comprising the sequence, interaction network, and metabolic model analyses, which would be an inception toward developing novel antibiotic agents.
Materials and Methods
The workflow utilized in the present study to predict essential proteins in K. pneumoniae is depicted in Figure 1.

Overview of the methodology adopted in the study comprising various approaches to predict essential proteins in Klebsiella pneumoniae. GSMM/GSSM, Genome-Scale Metabolic Model; PPI, protein–protein interaction.
Data retrieval
The complete reference proteome sequence of K. pneumoniae subsp. pneumoniae (strain ATCC 700721/MGH 78578) was retrieved from UniProtKB (Proteome ID: UP000000265) (UniProt Consortium, 2023). The protein–protein interaction (PPI) network data of K. pneumoniae was mapped using the retrieved whole proteome sequence, with a confidence score ≥0.900 and was imported into the Cytoscape workspace from the STRING database (v11) (Szklarczyk et al., 2019) using StringApp (Doncheva et al., 2019).
Network and cluster analysis
The mapped PPI network of K. pneumoniae was visualized in Cytoscape (version 3.8.2), and various network centrality measures were computed using NetworkAnalyzer, an in-built Cytoscape plugin for computing network topological metrics (Shannon et al., 2003). cytoHubba is a Cytoscape-based app that identifies essential nodes in the network and ranks those nodes based on different topological analysis methods (Chin et al., 2014). Cluster analysis of the constructed PPI network was performed using Molecular Complex Detection (MCODE), a Cytoscape app plugin (Bader and Hogue, 2003), and the enrichment of the clusters was carried out with StringApp with no redundant terms (p < 0.05).
Essentiality prediction
The present study predicted essential proteins of K. pneumoniae using an integrative bioinformatics and systems biology approach comprising the ensemble of sequence, PPI network, and GSSM methods.
Sequence-based prediction
Database of essential genes (DEG) is an online database that comprises experimentally identified set of essential genes/proteins of bacteria, archaea, and eukaryotes (Luo et al., 2014). The retrieved whole proteome sequence of Klebsiella pneumoniae MGH 78578 was searched against essential proteins of all bacteria present in the DEG 10 using BLASTp with the criteria of E value ≤1e−05 and query coverage ≥80%. If a protein sequence showed similarity to at least one hit from the database sequence with the above-mentioned criteria, then it was considered as an essential protein in the present study. In addition, the proteome sequence was subjected to essentiality prediction using Geptop 2.0, a prediction server for gene essentiality of prokaryotes (Wen et al., 2019).
PPI network-based prediction
The nodes of the constructed PPI network of K. pneumoniae were ranked based on network topological parameters using cytoHubba (11 parameters) and Network Analyzer (clustering coefficient) plugins. From each topological centrality parameter, the top 1000 nodes were considered and compared. If a protein node was present in at least two centrality parameters, it was considered as an essential protein from this analysis.
GSSM-based prediction
The GSSM (iLY1228) of Klebsiella pneumoniae MGH 78578 (Liao et al., 2011) present in the BiGG Models database (Norsigian et al., 2020) was analyzed using the MetaNetX platform, an interactive online resource for the automated construction, annotation, and analysis of large-scale metabolic networks (Moretti et al., 2021). A gene/peptide knockout (PKO) analysis was carried out on the iYL1228 model, which identified genes/peptides that hinder the growth of the organism.
Finally, if two or more prediction approaches predicted a protein to be essential, then it was considered as an essential protein in this study. This set of essential proteins was compared with NetGenes, an online database comprising machine learning-based prediction of essential genes in 2711 bacteria (Senthamizhan et al., 2021). Further, the expression of these essential proteins was explored using the PATHOgenex platform, a comprehensive RNA atlas of global expression profiles of 32 human pathogens under stress conditions (Avican et al., 2021).
Results and Discussion
Proteome of K. pneumoniae and interactome data
The complete proteome sequence of K. pneumoniae subsp. pneumoniae MGH 78578 comprises 5126 proteins encoded in a single circular chromosome with five plasmids (pKPN3, pKPN4, pKPN5, pKPN6, pKPN7). Out of 5126 proteins, only 715 were under the reviewed category in UniProtKB, suggesting their extensive curation and annotation levels. The genome accessions and the protein count are provided in Supplementary Table S1. The PPI data from the STRING database for K. pneumoniae had 5797 distinct protein-coding genes belonging to the core STRING type. The PPI data of K. pneumoniae in the STRING database is based on the integrated data from various resources. This core set represents the most reliable and supported PPI data based on several evidences and biological relevance.
Upon mapping the retrieved sequence data to the PPI data of the STRING database, a total of 4447 proteins were mapped with an interaction score ≥0.900 (highest confidence). This reduction in the number of proteins mapped (4447 out of 5126) to the PPI data was due to the stringent cutoff of the confidence score, limited data/annotations and interactome complexity.
PPI network of K. pneumoniae
The mapped PPI data of K. pneumoniae, imported into Cytoscape, was analyzed with global and local network topological parameters. In the PPI network, the proteins are represented as nodes, and their interactions are represented as edges. The constructed PPI network of K. pneumoniae comprised 4447 nodes with 8897 interactions (Supplementary Fig. S1), with 7.6 as the average number of neighbors. The average number of neighbors refers to the average degree of connectivity between the proteins, that is, the average number of interacting partners each protein has in the network.
The average network degree can be computed by summing the degrees of each node divided by the network's total number of nodes. The degree of a node (protein) is defined as the number of edges (interactions) connected to the specific protein in the network. The degree suggests the importance of a node (protein), where proteins with high degrees are referred to as hubs, which play a central role in the network and are crucial for various biological functions (Zotenko et al., 2008).
On the contrary, proteins with a lower degree have fewer interactions that may involve in specific interactions for a specialized function within a cell. Analyzing the degree distribution in the PPI network can provide insights into the overall topology and structure of the network. It assists in identifying highly connected proteins that can act as key regulators or mediators in cellular processes and also in understanding how proteins interact to form functional modules or participate in a pathway. In addition, this also provides the robustness and vulnerability of the network for perturbations. The computed global network statistical properties of the constructed network are given in Table 1. The degree distribution of the constructed PPI network of K. pneumoniae (Fig. 2) exhibited a long-tail pattern following a power-law distribution, indicating the scale-freeness property of the network.
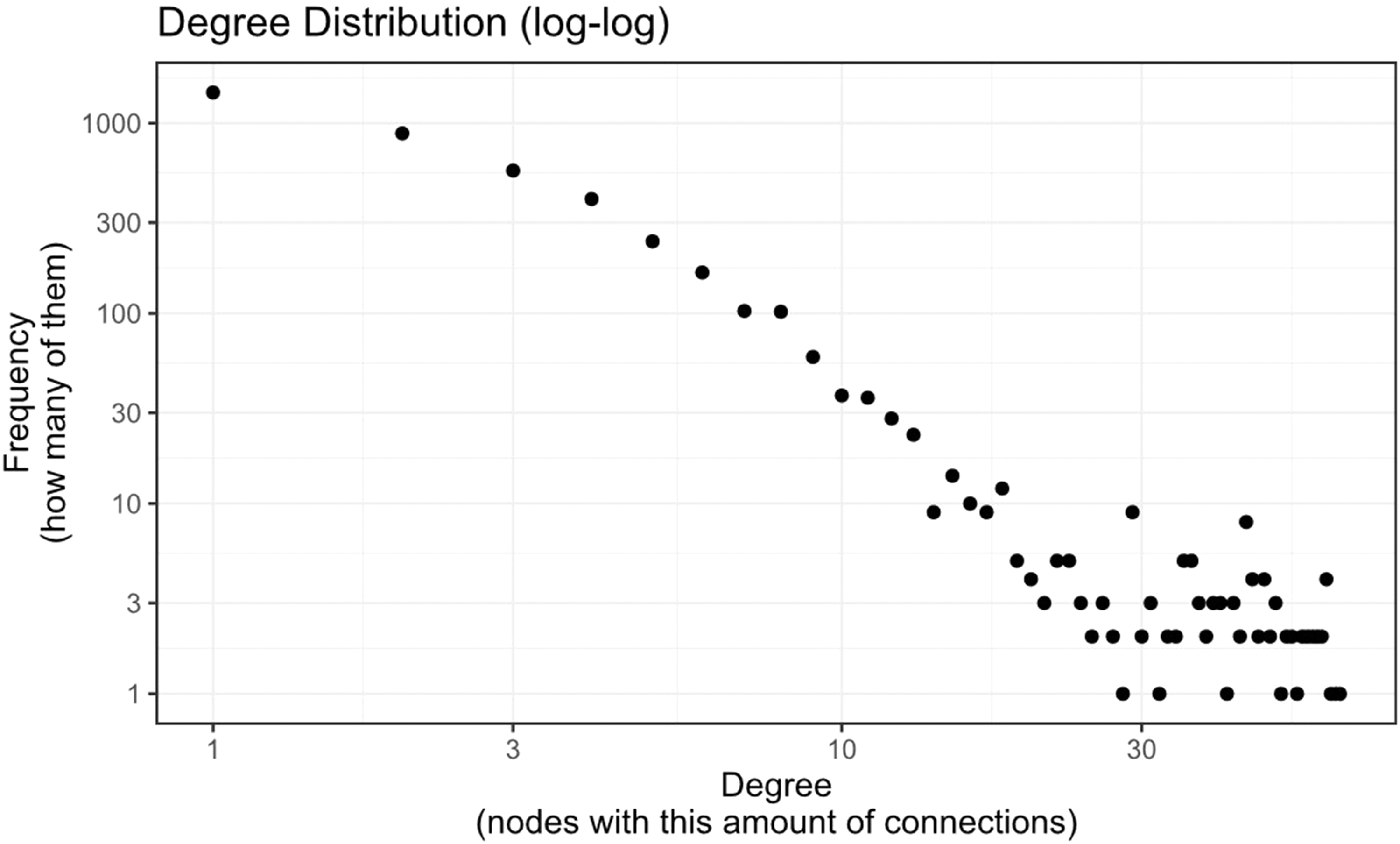
Scatter plot of the degree distribution of the constructed PPI network of Klebsiella pneumoniae.
Global Network Topology of the Constructed Protein–Protein Interaction Network of Klebsiella pneumoniae
The power-law distribution implies that a minimal number of nodes (proteins), referred to as hubs, have higher interactions (degree) than most of the proteins with lesser interactions. This signifies the network's resilience to perturbations, and the hubs act as crucial points that can potentially disrupt cellular functional regulations (Albert, 2005).
The network radius measures the shortest path between a specific node (protein) to the other farthest node, which shows the spread or dispersion of the network. The smaller the radius, the more compact and tightly interconnected the network is, where most of the proteins are closer to each other. Herein, the PPI network of K. pneumoniae had a network radius of eight, indicating that the traversal from a node (protein) had a path distance of eight edges to reach the farthest node to pass the information or signal. The identified characteristic path length for the K. pneumoniae PPI was 6.13. It indicates the average number of edges between nodes that exist in the network, showing the efficiency of information flow.
The clustering coefficient of the PPI network was 0.44, reflecting the local interconnectedness among the nodes (44%) in the network. The higher clustering coefficient suggests the existence of tightly connected complexes in the network. Several attributes, such as data availability, experimental techniques, and functional specificity, contribute to the representation of networks, which could be the limiting factors in network analysis.
The analysis of the global properties of the K. pneumoniae PPI network reveals insight into various important structural and functional properties of the network, such as scale-freeness, hubs, information flow, and connectedness. The scale-free topology indicates the network's resilience to perturbation, signifying the role of hubs in regulating cellular processes that are potentially essential to the organism. These findings enhance our understanding of K. pneumoniae protein interactions and their functional and organizational implications. In addition, it could shed light on potential key regulators or pathways for further exploration to combat infections and develop therapeutic strategies.
Cluster and enrichment analysis
The cluster analysis of the constructed PPI of K. pneumoniae resulted in 211 clusters with a minimum degree of >2. It showed the densely connected modules with significant functional relationships and likely to be involved in a common biological pathway or process. The 211 clusters indicate that the PPI network is modular with distinct functional units. Each identified cluster had a specific biological process, functional module, or protein complex. The top 10 clusters (Supplementary Fig. S2), identified based on the MCODE analysis and scores, are listed in Table 2. Enrichment analysis of these modules revealed the gene ontology terms related to the clusters and the pathways associated with the cellular processes.
Top 10 Cluster Scores Based on MCODE Analysis of the Constructed Protein–Protein Interaction Network of Klebsiella pneumoniae with the Number of Nodes and Edges
MCODE, Molecular Complex Detection.
The top three clusters with the highest scores are shown with STRING-based representations in Figure 3. Cluster 1 consisted of 58 nodes with 1615 edges, mostly ribosomal proteins (L13, S20, L2, L9, S17, L35, etc.) involved in rRNA (ribosomal RNA) or tRNA (transfer RNA) binding. Ribosomes are essential cellular organelles responsible for protein synthesis and comprise both rRNA and ribosomal proteins. This cluster 1 showed the ribosomal complexes, including the subunits forming functional roles to perform translation, an essential process in cellular functioning. Moreover, the dominance of ribosomal proteins in this cluster implies the role of these proteins in cellular regulation, homeostasis, and growth. In this cluster, ribosome, RNA polymerase, and protein export were found to be significantly enriched KEGG pathways, and nucleic acid binding (oligonucleotide/oligosaccharide-binding fold), zinc-binding ribosomal protein, beta-barrel domain, translation protein SH3-like domain superfamily and ribosomal protein S5 domain 2-type fold were enriched terms in InterPro domains.

Top 3 clusters of the Klebsiella pneumoniae protein–protein interaction network showing various nodes (proteins) interacting as functional modules associated with different vital biological processes.
Cluster 2 had 18 nodes with 152 edges that were enriched with proteins involved in cobalamin biosynthesis, including the tetrapyrrole methylase, CbiD, CbiC, CobU, CbiF, CbiH, CbiO, and so on, which signifies a functionally cohesive group of proteins involved in cobalamin biosynthesis. It has been reported that CbiD is essential for cobalamin biosynthesis in both Salmonella typhimurium and Bacillus megaterium (Raux et al., 1998). It is known that bacteria synthesize cobalamin, which is an essential coenzyme for vital cellular processes such as nucleic acid metabolism and amino acid synthesis. This cluster signifies the interactions between proteins coordinated in cobalamin synthesis, which is important for bacterial growth and metabolism. The pathway enrichment of cluster 2 showed significant pathways that the proteins were involved in, namely pyruvate metabolism, carbon metabolism, biosynthesis of secondary metabolites, and propanoate metabolism, which are essential for cellular regulation and metabolism.
Similarly, cluster 3 was observed to have 16 nodes with 117 edges, comprising mainly proteins of nicotinamide adenine dinucleotide (NAD) + hydrogen (H)-quinone (NADH-quinone) oxidoreductase subunits (NuoA, NuoB, NuoC, NuoH, NuoI, NuoN, and NuoK, etc.), enriched in ubiquinone and peptidase M16 activity with NADH-quinone oxidoreductase/Mrp antiporter domains, indicating potential role in substrate or ion transport across membranes. The NADH-quinone oxidoreductase complex has been reported to have a key role in bacterial respiration for ATP generation (Friedrich et al., 2016). The protein peptidase M16 belongs to the metallopeptidase family and has diverse functional roles, including proteolytic activity, that are important for protein processing and degradation (Turner and Nalivaeva, 2011). The proteins of this cluster were mainly involved in electron transport and energy production for cellular respiration. The presence of ATP-binding cassette transporters in the identified cluster indicates their involvement in nutrient uptake and metabolite efflux in the bacterial cell for maintaining homeostasis (Davidson et al., 2008).
The pathway enrichment analysis of the cluster showed that the proteins were mainly involved in porphyrin metabolism and transport. Porphyrin metabolism is associated with heme synthesis, which is important for several cellular processes such as electron transport, oxygen binding, and so on (Dailey et al., 2017).
The cluster analysis with enrichment of the modules identified from the PPI network unveiled the modularity property of the network involved in distinct functions/processes. The top three clusters comprised proteins associated with various essential functions such as translation, cobalamin biosynthesis, energy metabolism, and transport. These findings highlight the functional organization of clusters in the PPI network belonging to biological pathways and processes that are central to the cellular functioning of K. pneumoniae.
Predicted essential proteins of K. pneumoniae
The prediction of essential genes/proteins in an organism aids in identifying vital factors that are essential to the organisms' survival and thus can be considered as drug target candidates. These factors can be utilized for the development of drugs with selective binding to avoid any side effects on the host. In addition, they also help in understanding the basic core cellular functional regulations, which have broad applications in the area of applied biotechnology and biomedicine (Dong et al., 2020). The current study utilized a combination of ensemble approaches comprising integrative systems biology and bioinformatics methods to predict the essential proteins in K. pneumoniae.
The sequence-based prediction identified essential proteins based on similarity to existing experimentally determined essential proteins presented in the DEG database. The concept in this similarity-based method is that proteins that have a high similarity to known essential proteins of other organisms/pathogens are potentially essential as they could be homologs or orthologs in nature. These proteins are often found to be highly conserved across different closely related species, maintaining functional conservation that is essential to basic cellular functionality, which can assist in studying evolution and biological functional conservation.
The sequence-based approach resulted in 744 K. pneumoniae proteins that were similar to essential proteins identified in various bacterial strains of Escherichia coli, Salmonella enterica, Providencia stuartii, Shewanella oneidensis, Pseudomonas aeruginosa, Vibrio cholerae, Haemophilus influenzae, and so on. Thus, these 744 proteins are likely to be essential in K. pneumoniae. These proteins belong to the gram-negative class of bacteria, indicating their conservation across different species. For instance, the ribosomal proteins and subunits (large and small) identified as essential had high similarity (≈100%) and are known to be highly conserved across bacterial species (van den Elzen et al., 2023; Lecompte et al., 2002).
Other proteins identified based on the similarity approach are peptidyl-tRNA hydrolase, recombinase A, dihydroorotase, Ribonuclease 3, pyridoxine/pyridoxamine 5′-phosphate oxidase, queuine tRNA-ribosyltransferase, and so on. These proteins were found to be involved in major essential processes such as tRNA processing, translation, DNA recombination, ribosomal small subunit biogenesis, RNA catabolic process, regulation of DNA-templated transcription, nicotinamide dinucleotide phosphate (NADP) metabolic process, and cell wall organization, including lipid A biosynthetic process and peptidoglycan biosynthetic process. These processes are vital for regular cellular functionality, and these proteins signify their indispensability potential for further investigations toward drug development. Since the similarity-based approach is highly dependent on the conservation and annotations of gene/protein information, it relies on the availability of experimental data.
In this context, prediction-based methods using the sequence data could provide reliable predictions at the genome scale for gene essentiality. One such tool is Geptop 2.0, based on public essential gene repositories for identifying essential genes in prokaryotes. This tool predicted 394 proteins as essential proteins in K. pneumoniae. For example, the proteins UDP-N-acetylmuramate—L-alanine ligase (encoded by murC), small ribosomal subunit protein uS4 (rpsD), phenylalanine—tRNA ligase alpha subunit (pheS), uridylate kinase (pyrH), and elongation factor Ts (tsf) were some of the proteins with high essentiality scores. Geptop classifies a protein as essential (1) or non-essential (0) based on the essentiality scores defined by a cumulative formula and are normalized to a range from zero to one. The larger the value toward one represents the essentiality of the gene/protein, and the average area under the curve (AUC) is ∼0.84, as reported (Wen et al., 2019).
Functional analysis of the Geptop predicted essential proteins showed these proteins were majorly involved in key biological processes, including translation, peptidoglycan biosynthesis, ATP binding, rRNA binding, lipoprotein biosynthesis, methionyl-tRNA aminoacylation, and so on. The results of the prediction-based method were in accordance with the similarity-based approach in revealing the core essential biological processes in bacteria, indicating functional significance.
In addition to the sequence-based approach (similarity and prediction), using the constructed PPI network of K. pneumoniae, a network centrality-based approach was utilized to rank the proteins to shortlist essential proteins using 11 topological parameters computed based on cytoHubba along with another topological parameter, clustering coefficient. The correlation plot based on these 12 topological parameters (degree, Edge Percolated Component, Maximum Neighborhood Component, Density of Maximum Neighborhood Component, Maximal Clique Centrality, bottleneck, eccentricity, closeness, radiality, betweenness, stress, and clustering coefficient) is depicted in Supplementary Figure S3, showing the relationship between computed values.
Based on these 12 topologies, the protein nodes of the PPI network were ranked (Top 1000), and a protein was considered as essential if it is present in at least two centrality measures. This PPI-based prediction resulted in a total of 1131 proteins, which were found to possess central properties in the network and perturbation of such proteins would collapse the network and possibly be essential in K. pneumoniae.
Some of the proteins identified as essential based on the network topologies were ribosomal proteins (30S S9, 30S S5, 50S L15, 50S L6, 50S L2, etc.), translation elongation factor LepA, DNA-directed RNA polymerase, translation initiation factor IF-3, cobalamin biosynthesis protein CbiG, threonine—tRNA ligase, NADH-quinone oxidoreductase subunit C, pyruvate-flavodoxin oxidoreductase, Type II secretion system proteins (J, I, L, G, K), CoA-acylating propionaldehyde dehydrogenase, and so on. The protein pyruvate-flavodoxin oxidoreductase was identified as the highest interaction protein node with a degree of 82 in the PPI network of K. pneumoniae, a crucial enzyme for metabolic repertoire that allows bacteria to adapt and survive in different environmental conditions. This network-based approach provides a unique way to integrate data resources in the context of PPIs to assess functional relationships, which is crucial in essentiality, but it is limited to data quality, availability, and completeness.
Overall, these network topologies provide reliable metrics to pinpoint essential proteins as they denote their central roles and contribution toward network functionality and integrity, enhancing confidence in the prediction of essentiality. However, combining multiple topologies and integrating them with experiments is necessary to validate these predictions.
Further, a GSSM iYL1228 of K. pneumoniae was subjected to gene/PKO analysis, resulting in 118 peptides that critically affected biomass production and were lethal when knocked out. These 118 peptides/proteins are predicted to be essential in the analyzed GSSM of K. pneumoniae. This experimentally validated metabolic model comprises 1229 genes with 1658 metabolites involved in 2262 reactions. Cytidine 5-triphospate (CTP) synthase [EC (Enzyme Commission number) 6.3.4.2], orotidine 5′-phosphate decarboxylase (EC 4.1.1.23), phosphatidylserine synthase, acyl carrier protein, tryptophan synthase alpha chain (EC 4.2.1.20), ATP phosphoribosyltransferase (EC 2.4.2.17), histidine biosynthesis bifunctional protein HisB, and UDP-glucose 6-dehydrogenase were some of the identified essential genes/proteins that crucially affect the growth.
It was observed that these essential proteins were part of crucial biosynthetic processes such as lipid A biosynthesis, carbohydrate metabolism, amino acid biosynthesis, phosphorylation, polysaccharide biosynthesis, fatty acid biosynthesis, de novo inosine monophosphate/adenosine monophosphate (IMP/AMP) biosynthesis, transsulfuration, chorismate metabolic process, and so on. These results can assist in designing and developing tools such as metabolic engineering for various applications and potentially enhance our understanding of complex metabolic networks that are fundamental to life forms.
To obtain a comprehensive set of essential proteins in K. pneumoniae, proteins were compared between different prediction approaches, and it was found that 33 proteins (Fig. 4; Table 3) were common in all prediction approaches. Herein, the final set of essential proteins was considered based on the presence of a protein in two or more prediction approaches. A total of 854 proteins (Supplementary Table S2) were identified as the consensus set of essential proteins based on the results of the combination approach. These 854 proteins can be further evaluated experimentally to validate the findings from the study.
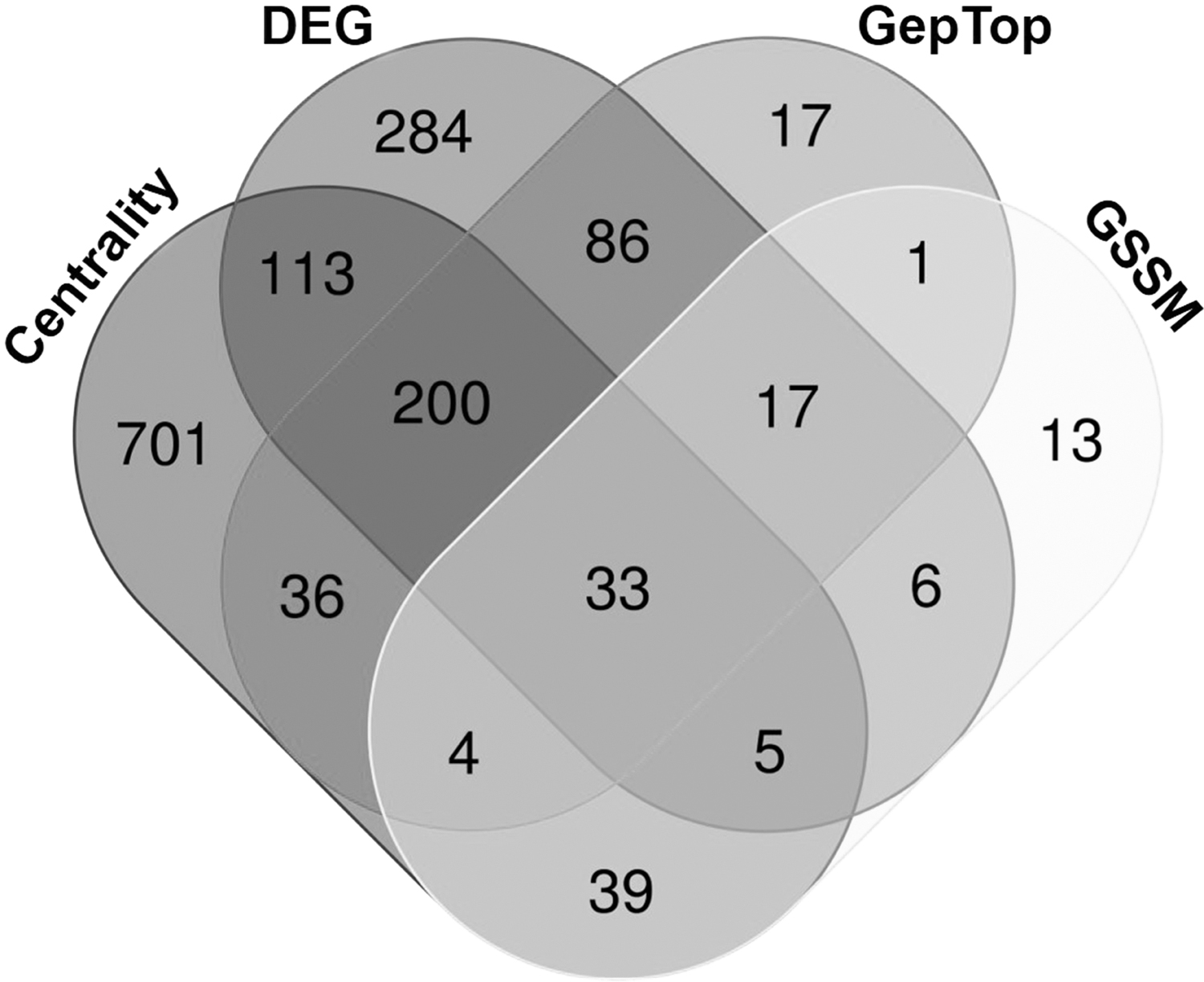
Venn diagram showing the number of essential proteins overlapping with different prediction approaches. DEG, database of essential genes; GSSM, genome-scale metabolic model.
List of 33 Klebsiella pneumoniae Proteins Predicted as Essential By All Methods in the Present Study
AA, amino acids; PDB, Protein Data Bank.
In silico validation of predicted essential proteins
The final comprehensive set of 854 essential proteins predicted based on the consensus combination approach was compared with the NetGenes database, which utilized network-based features to predict essential proteins from the STRING PPI data. It was noticed that 242 proteins (Fig. 5) were common among the essential proteins predicted from the present study, with NetGenes essential proteins showing the validity of the approach utilized. A recent study that curated a set of Klebsiella metabolic models proved 57 genes to be essential for all substrates in all considered strains (Hawkey et al., 2022), and interestingly, all those 57 genes/proteins from the present study were also identified as essential.
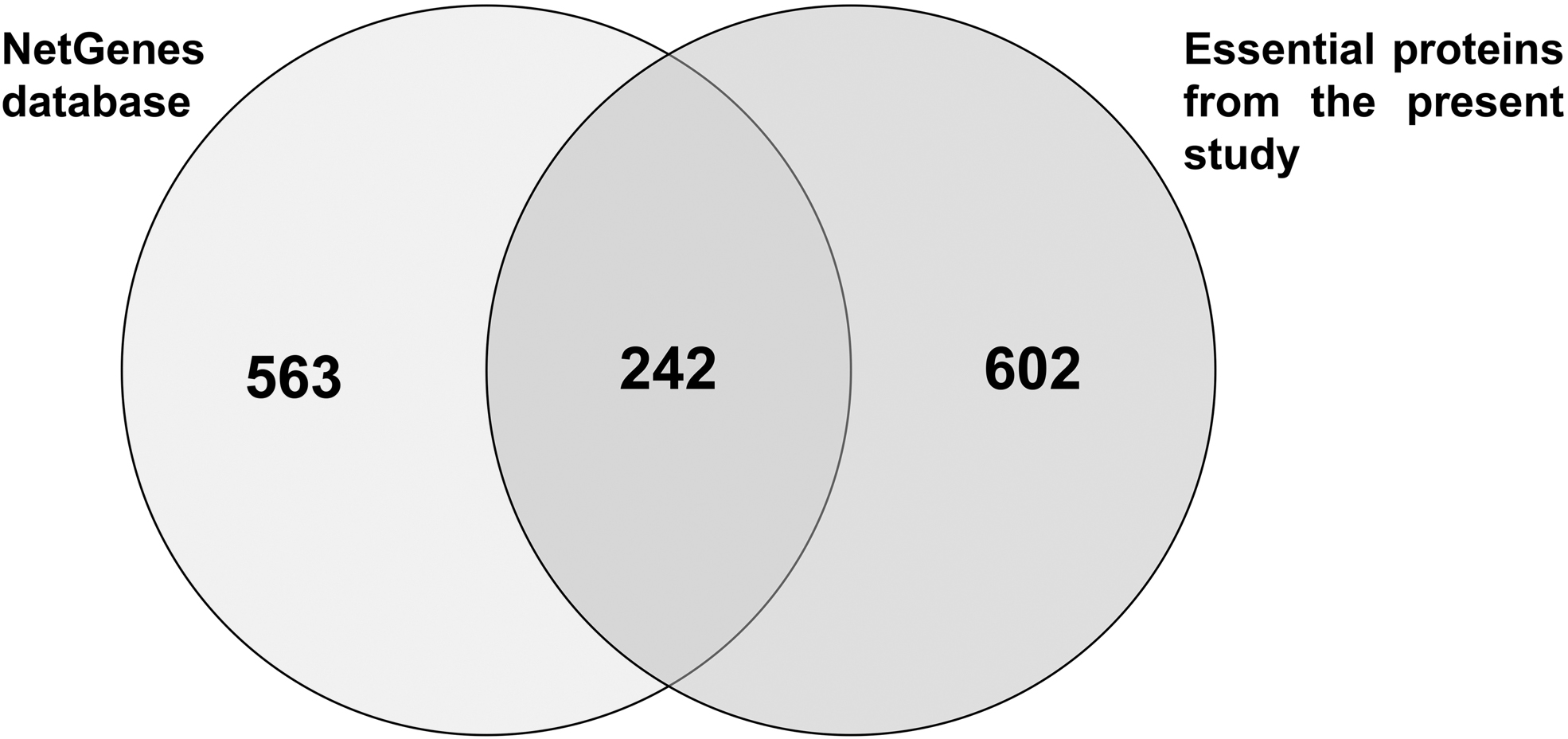
Venn diagram showing the number of common genes/proteins predicted as essential of Klebsiella pneumoniae in the present study and essential proteins of the NetGenes database.
In addition, the gene expression of the predicted essential proteins was explored with the PATHOgenex platform, a resource for human bacterial pathogens gene expression data (RNA atlas). It was observed that these essential proteins were differentially expressed in different conditions such as acidic stress (As), bile stress (Bs), low iron (Li), hypoxia (Hyp), nutritional downshift (Nd), nitrosative stress (Ns), osmotic stress (Oss), oxidative stress (Oxs), stationary phase (Sp), temperature (Tm), and virulence inducing condition (Vic) (Supplementary Fig. S4). The condition-specific expression of these essential proteins suggests that these proteins are associated with the adaptiveness of the pathogen in different environments. In particular, the essential proteins that are expressed in the Vic condition have a potential association with pathogenicity and thus can be considered as target candidates for developing antimicrobial agents specific to them.
Overall, the findings of this study can contribute to our fundamental understanding of essentiality and cellular functions that can, in turn, lead to target-based drug discovery and development to tackle infectious diseases caused by bacterial pathogens.
Conclusions
The prediction of essential genes/proteins has several applications in biology and medicine. It helps to comprehend the core fundamental biological processes vital for an organism's survival. Identifying essential proteins also guides toward a targeted drug discovery approach by shortlisting potential therapeutic targets that could facilitate the development of novel antimicrobial agents against pathogenic infections. The present study utilized an integrative bioinformatics and systems biology approach to identify essential proteins in K. pneumoniae. The combination approach of proteome sequence, interaction, and genome-scale modeling methods resulted in 854 essential proteins, involved in major cellular and biological processes. Cluster analysis of the PPI network of K. pneumoniae showed the top regulatory pathways and processes that are vital for cellular functions. The predicted essential proteins were compared with existing databases and literature that confirmed the validity of the prediction approach.
The findings of the study would serve as a beginning for a target-based drug discovery approach to consider these essential proteins for shortlisting novel antimicrobial agents toward therapeutic development. Moreover, the methodology adopted herein can be exploited and applied to other multidrug-resistant pathogens that cause infections to humankind.
Footnotes
Acknowledgments
G.P. is supported by the Department of Biotechnology (DBT), Government of India, under the DBT-BINC research fellowship program (DBT-BINC/2017/PU/6). The authors are indebted to the Department of Bioinformatics, Pondicherry University, Pondicherry, for providing the computational facility to carry out the work.
Authors' Contributions
G.P.: Conceptualization, methodology, data analysis, visualization, and writing—original draft, review, and editing. A.P.: Conceptualization, formal analysis, resources, supervision, and writing—review and editing. All authors have read and approved the final version of the article.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
No funding was received for this article.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.