
Abstract
Cerebral vasospasm (CV) is a significant complication following aneurysmal subarachnoid hemorrhage (aSAH), and lacks a comprehensive molecular understanding. Given the temporal trajectory of intracranial aneurysm (IA) formation, its rupture, and development of CV, altered gene expression might be a molecular substrate that runs through these clinical events, influencing both disease inception and progression. Utilizing RNA-Seq, we analyzed tissue samples from ruptured IAs with and without vasospasm to identify the dysregulated genes. In addition, temporal gene expression analysis was conducted. We identified seven dysregulated genes in patients with ruptured IA with vasospasm when compared with those without vasospasm. We found 192 common genes when the samples of each clinical subset of patients with IA, that is, unruptured aneurysm, ruptured aneurysm without vasospasm, and ruptured aneurysm with vasospasm, were compared with control samples. Among these common genes, TNFSF13B, PLAUR, OSM, and LAMB3 displayed temporal expression (progressive increase) with the pathological progression of disease that is formation of aneurysm, its rupture, and consequently the development of vasospasm. We validated the temporal gene expression pattern of OSM at both the transcript and protein levels and OSM emerges as a crucial gene implicated in the pathological progression of disease. In addition, RSAD2 and ATP1A2 appear to be pivotal genes for CV development. To the best of our knowledge, this is the first study to compare the transcriptome of aneurysmal tissue samples of aSAH patients with and without CV. The findings collectively provide new insights on the molecular basis of IA and CV and new leads for translational research.
Introduction
Cerebral vasospasm (CV) is one of the potentially fatal complications occurring in patients surviving the aneurysmal subarachnoid hemorrhage (aSAH). The angiographic vasospasm is seen approximately in 70% of patients with aSAH, whereas clinical symptoms develop in 30% of the patients (Khanafer et al., 2022). Clinically symptomatic vasospasm causes reduced blood flow to the affected area of the brain, leading to ischemic brain injury or ischemic stroke. The incidence rate of 20% for both mortality and morbidity is described in those affected by this syndrome (Khanafer et al., 2022). Various pharmacological and medical interventions have been attempted for the prevention and treatment of CV (Chan et al., 2021) without an appreciable effect to overall modify the clinical outcome.
Despite the various hypotheses on the pathophysiology of CV, its molecular mechanism is still poorly understood. Over the past several decades, investigation of the genetic bases of CV has been a leading frontier of research and is also supported by previous population studies. Molecular genetics has led to the identification of candidate genes and their products, which could be the mediators of CV (Ateia et al., 2020; Ladner et al., 2013; Wu et al., 2010). Various genetic variants that envisage an increased risk of CV or poor outcome have also been revealed (Medina-Suárez et al., 2022; Solodovnikova et al., 2021). However, these findings have not been translated to new interventions or diagnostics that can markedly reduce the disease burden.
Omics technologies such as transcriptomics and the high-throughput study of gene expression offer the promise of uncovering the dysregulated genes, biological processes, and pathways that underpin CV. We designed the present study to understand the role of altered gene expression in the development of CV after aSAH. Previously, several gene expression studies have been done using blood samples (Pulcrano-Nicolas et al., 2019; Sasahara et al., 2008; Xu et al., 2020). However, to the best of our knowledge, this is the first study to compare the transcriptome of aneurysmal tissue samples of patients with aSAH with and without CV.
The present study follows in continuation of our previous study in which we performed the transcriptomic analysis of unruptured intracranial aneurysm (IA) and ruptured IA (without vasospasm) tissue samples along with controls to identify the dysregulated genes (Kumar et al., 2023). In the current study, we acquired the transcriptomic data of tissue samples of ruptured IA with vasospasm and compared them with the transcriptomic data of tissue samples of ruptured IA without vasospasm (acquired in the previous study) to identify the dysregulated genes specific for the development of vasospasm following aSAH. Moreover, we performed the temporal gene expression analysis to identify the genes that were progressively upregulated or downregulated with the pathological progression of the disease, that is, the formation of the aneurysm, its rupture, and consequently the development of vasospasm.
Materials and Methods
Patient selection, sample collection, and storage
This study was conducted at Postgraduate Institute of Medical Education and Research (PGIMER), Chandigarh, India, in collaboration with the Institute of Bioinformatics, Bangalore, India. The Institute Ethics Committee of PGIMER (IEC No. MK/2866/Ph.D/7735, Dated: October 4, 2016) has approved this study. The subjects were recruited from the Departments of Neurosurgery, Neurology, and Forensic Medicine at PGIMER after obtaining written informed consent from patients/relatives. The patient population consisted of subjects 18–65 years diagnosed/confirmed with unruptured IA, ruptured IA without vasospasm, and ruptured IA with vasospasm using computed tomography scan and angiographic examination. The subjects with normal/healthy vasculature on angiographic examination or autopsy were included in the control group. The samples for control, unruptured IA, and ruptured IA without vasospasm groups were collected as described previously (Kumar et al., 2023).
The control tissue samples (intracranial arterial tissue) were collected from the subjects undergoing autopsy, while blood samples were collected from the subjects visiting the neurology clinic. Aneurysm tissue samples for each group (unruptured IA, ruptured IA without vasospasm, and ruptured IA with vasospasm) were obtained intraoperatively after the aneurysm clipping.
Furthermore, to minimize the blood contamination, tissue samples were rinsed with normal saline and incubated overnight in RNAlater at 4°C. The next day, tissue samples were transferred in a fresh RNAlater vial and stored at −80°C until further use. Blood samples were collected from the aneurysm patients before surgery in plain vials. Furthermore, blood samples were allowed to coagulate in normal conditions and centrifuged to obtain serum (supernatant). All the processes were carried out in accordance with the Institute's rules and regulations.
Data availability
The data sets generated for this study can be found in the Sequence Read Archive hosted by the National Center for Biotechnology Information search database (NCBI) with accession number: PRJNA524023.
RNA sequencing and data analysis
The RNA sequencing and data analysis were performed as described in our previous study (Kumar et al., 2023). In brief, total RNA was extracted from tissue samples followed by RNA quantification and integrity analysis. Furthermore, 500 ng of total RNA from each sample was subjected to ribodepletion. Illumina TruSeq RNA Prep Kit V2 (Illumina, San Diego, CA, USA) was used for library preparation. Cluster generation was done using the Illumina cBot system, and the Illumina HiSeq 2500 platform (Illumina) was used for sequencing. The paired-end reads of length 100 bp (2 × 100) were generated with a library size of 30–58 million reads.
FASTQC (version 0.11.2) tool was used for the quality filtration of raw reads obtained in fastq format (Andrews, 2010). Furthermore, low-quality ends having a Phred quality score Q < 30 were trimmed off using Fqtrim (version 0.94) (Pertea, 2015). For the downstream analysis, reads with length ˃70 bps were retained. Filtered, high-quality reads were aligned to reference genome HG38 (GENCODE version 26) using the HISAT2 (version 2.0.1) (Kim et al., 2015). The StringTie (version 1.2.1) was used to assemble aligned reads against the reference genome (GRCH38) (Pertea et al., 2015). Transcripts were aligned and assembled separately for each sample and read counts were obtained, which were further normalized. The DESeq package was used for differential gene expression analysis (Anders and Huber, 2010).
We investigated only those genes that were quantified with ≥30 reads in at least three samples per cohort. Correction for false discovery rate (FDR) of 5% was made using the Benjamin–Hochberg procedure. Genes with FDR-adjusted p-value of ≤0.05 and log2fold change cutoff (≥2 and ≤ −2) were considered significantly dysregulated.
Functional and pathway analysis
Volcano plots were generated using the R package to visualize the results of the differential expression analysis (Supplementary Fig. S1). Heatmap was created using Morpheus software (https://software.broadinstitute.org/morpheus/). In addition, GeneCodis4 was used for Gene Ontology (GO) enrichment analysis of dysregulated genes (Garcia-Moreno et al., 2022). For pathway analysis, both DAVID and GeneCodis4 software were utilized (Huang da et al., 2009). To delve into the function of dysregulated genes and construct the protein–protein interaction (PPI) network, the STRING tool (version 11.0) was used (Szklarczyk et al., 2019). The obtained interaction networks were exported to Cytoscape (version 3.6.0) for visualization (Shannon et al., 2003). The cluster analysis was performed using MCODE plugin software in Cytoscape. Clusters with the higher number of nodes were subjected to further analysis. A p-value of ≤0.05 was considered significant for enriched GO terms and pathways.
Candidate genes were short-listed based on statistical significance, fold change, molecular function, and biological relevance, and subsequently selected for validation.
Temporal gene expression analysis
We conducted the temporal gene expression analysis to discern the genes exhibiting progressive upregulation or downregulation during the pathological progression of the disease, that is, formation of aneurysm (unruptured IA), its rupture (ruptured IA without vasospasm), and the subsequent development of vasospasm (ruptured IA with vasospasm). Venn diagrams were constructed to identify genes that were commonly differentially expressed across the groups. The significantly differentially expressed genes were used for Venn diagram generation.
Validation
We performed the validation at both transcript and protein levels using quantitative reverse transcription-polymerase chain reaction (qRT-PCR) and enzyme-linked immunosorbent assay (ELISA), respectively. An independent set of samples was utilized for the validation. Sample processing and further qRT-PCR and ELISA were performed as described in our previous study (Kumar et al., 2023). Briefly, total RNA was extracted from tissue samples using the TRIzol reagent, followed by cDNA synthesis. Subsequently, quantitative real-time PCR was performed to quantify the expression of candidate genes. Primers for candidate genes were designed using Primer-BLAST (Ye et al., 2012). For the validation at the protein level, serum samples were used. Precoated ELISA kits were used for the validation. Statistical analysis of data was performed using GraphPad Prism software version 6.01 (GraphPad Software, San Diego, CA, USA) and Microsoft Excel 2010.
An unpaired, Mann–Whitney test (nonparametric test) was used for assessing the statistical significance between groups, considering a p-value of ˂0.05 as statistically significant.
Results
To identify the dysregulated genes associated with CV, we performed the transcriptome analysis of six samples of ruptured IA without vasospasm (acquired previously) and five samples of ruptured IA with vasospasm (acquired in the present study). The clinicopathological details of the participants are given in Supplementary Table S1. The mean RNA integrity number for six samples of ruptured IA without vasospasm was 7.2 (6.7–8.5), while 7.08 (4.7–8.6) for five samples of ruptured IA with vasospasm (Supplementary Table S2). We generated more than 33 million high-quality reads (33–58 million) in each sample. The average read length was 100 bp. For 11 samples, the average Phred quality score (Q) was 39.14. More than 90% of reads (90.72–95.89%) in each sample were with Q ≥ 30, which showed the high quality of sequencing data. The reads with length <70 bps were excluded from the analysis.
Therefore, we remained with more than 28 million clean reads (28–51 million reads) in each sample for further downstream analysis. These high-quality, clean reads having length ≥70 bases were further aligned to reference genome Hg38 using HISAT2, keeping Gencode V26 as a backend annotation. We achieved ≥80% (80–93%) read alignment for each sample. Furthermore, we identified more than 11,000 genes in each sample, which were supported by ≥30 reads. A total of 13,888 genes were queried for differential expression analysis between these 2 subsets of patients with IA. Out of which only seven genes (protein coding) were found significantly differentially expressed (p ≤ 0.05 and log2FC ≥2 or ≤ −2). All differentially expressed genes were found to be upregulated in ruptured IA with vasospasm (Table 1).
Differentially Expressed Genes Between Ruptured Intracranial Aneurysm Without Vasospasm and Ruptured Intracranial Aneurysm with Vasospasm
Temporal gene expression analyses
For the temporal gene expression analysis, we utilized the transcriptome data of a cohort of 20 samples comprising 5 control samples, 4 samples of unruptured IA, 6 samples of ruptured IA without vasospasm, and 5 samples of ruptured IA with vasospasm. The transcriptome data of the samples of controls, unruptured IAs, and ruptured IAs without vasospasm were acquired in our previous study (Kumar et al., 2023), while RNA sequencing of five samples of ruptured IA with vasospasm was performed in the present study and transcriptome data were acquired. The clinicopathological details of the 20 participants are given in Supplementary Table S3. Significantly differentially expressed genes were considered for temporal expression analysis. We compared the control samples with unruptured IA samples and found 314 differentially expressed genes. Similarly, we compared the samples of unruptured IA with those of ruptured IA without vasospasm and found 301 differentially expressed genes (Kumar et al., 2023).
Furthermore, we found seven differentially expressed genes between ruptured IA without vasospasm and ruptured IA with vasospasm samples. We generated the Venn diagram to find the common genes. However, we did not find common significantly differentially expressed genes between these groups.
Furthermore, we compared the samples of each clinical subset of patients with IA, that is, (1) unruptured IA, (2) ruptured IA without vasospasm, and (3) ruptured IA with vasospasm independently with control samples. Subsequently, we identified 314 genes in unruptured IA versus control, 1415 genes in ruptured IA without vasospasm versus control, and 1233 genes in ruptured IA with vasospasm versus control that were significantly differentially expressed. Next, we generated the Venn diagram and found 192 differentially expressed genes that were common (Fig. 1a) in these 3 comparisons. Out of these 192 genes, 159 were upregulated and 33 were downregulated.

Functional analysis of common genes:
In the functional analysis of these common genes, we observed the enrichment for biological processes' immune response and antigen processing and presentation (Fig. 1c). We also found the enrichment for GO terms major histocompatibility complex (MHC) class II receptor activity, MHC class II protein complex binding, and peptide antigen binding under the molecular function-based classification (Fig. 1d). Furthermore, the pathway analysis revealed that these genes were involved in the pathways phagosomes, antigen processing and presentation, and cell adhesion molecules (Fig. 2a). The phagosome pathway is represented in Supplementary Figure S2.

Next, we performed the PPI network analysis of these 192 common genes. The PPI network thus obtained contains 182 nodes and 198 edges representing 192 genes (Supplementary Fig. S3). The average node degree was 2.18. A high confidence score threshold of value >0.7 was applied for the PPI network. Furthermore, we performed the cluster analysis. The clusters with more than five nodes were selected for analysis. We found two clusters having more than five nodes (Fig. 1e, f). Cluster 1 contains 20 nodes and 90 edges, while cluster 2 contains 7 nodes and 10 edges.
Furthermore, we obtain the expression values (log2fold change) for these common genes from the above three comparisons and screen for any progressive increasing or decreasing trend in their expression with the pathological progression of the disease. We identified four genes, that is, TNFSF13B, OSM, LAMB3, and PLAUR, that showed a temporal increase in their expression level as the disease progressed from the development of aneurysm to its rupture and consequently the development of vasospasm (Fig. 2b–e). The details of their gene expression in different groups are represented in Table 2. The genes TNFSF13B and OSM were found to be associated with the GO term “cytokine activity,” whereas PLAUR and LAMB3 were related to “signaling receptor activity” and “structural molecule activity,” respectively.
Differentially Expressed Common Genes Between T1 Versus C, T2 Versus C, and T3 Versus C with Temporal Gene Expression
Expression in log2fold.
C, control; IA, intracranial aneurysm; T1, unruptured IA; T2, ruptured IA without vasospasm; T3, ruptured IA with vasospasm.
Validation
We validated the temporal expression trend of OSM at both the transcript and protein levels using qRT-PCR and ELISA, respectively. The validation was carried out in a large independent cohort of samples. For this purpose, a fresh cohort of patients/subjects was recruited as described in the section “Patient selection, Sample collection, and Storage.” We utilized unpaired tissue and serum samples for the validation phase. Therefore, a total of 135 confirmed IA patients were recruited and 40 IA tissue samples (7 unruptured IA, 22 ruptured IA without vasospasm, and 11 ruptured IA with vasospasm) and 95 serum samples (16 unruptured IA, 39 ruptured IA without vasospasm, and 40 ruptured IA with vasospasm) were collected from these patients. In addition, 34 subjects were recruited into the control group and 15 tissue and 19 serum samples were collected from these subjects.
The clinicopathological details of patients/subjects recruited for the validation phase are provided in Supplementary Table S4. The OSM was the candidate molecule in our previous study for the group unruptured IAs versus controls and was validated at both the transcript and protein levels. The validation data of OSM at the transcript level from our previous study (Kumar et al., 2023) have been utilized in the present study. GAPDH was used as a housekeeping gene for normalization in qRT-PCR. We observed progressive upregulation in the expression of OSM at both the transcript and protein levels during the pathological progression of the disease, as each disease condition was individually compared with the control (Fig. 3 and Supplementary Table S5).
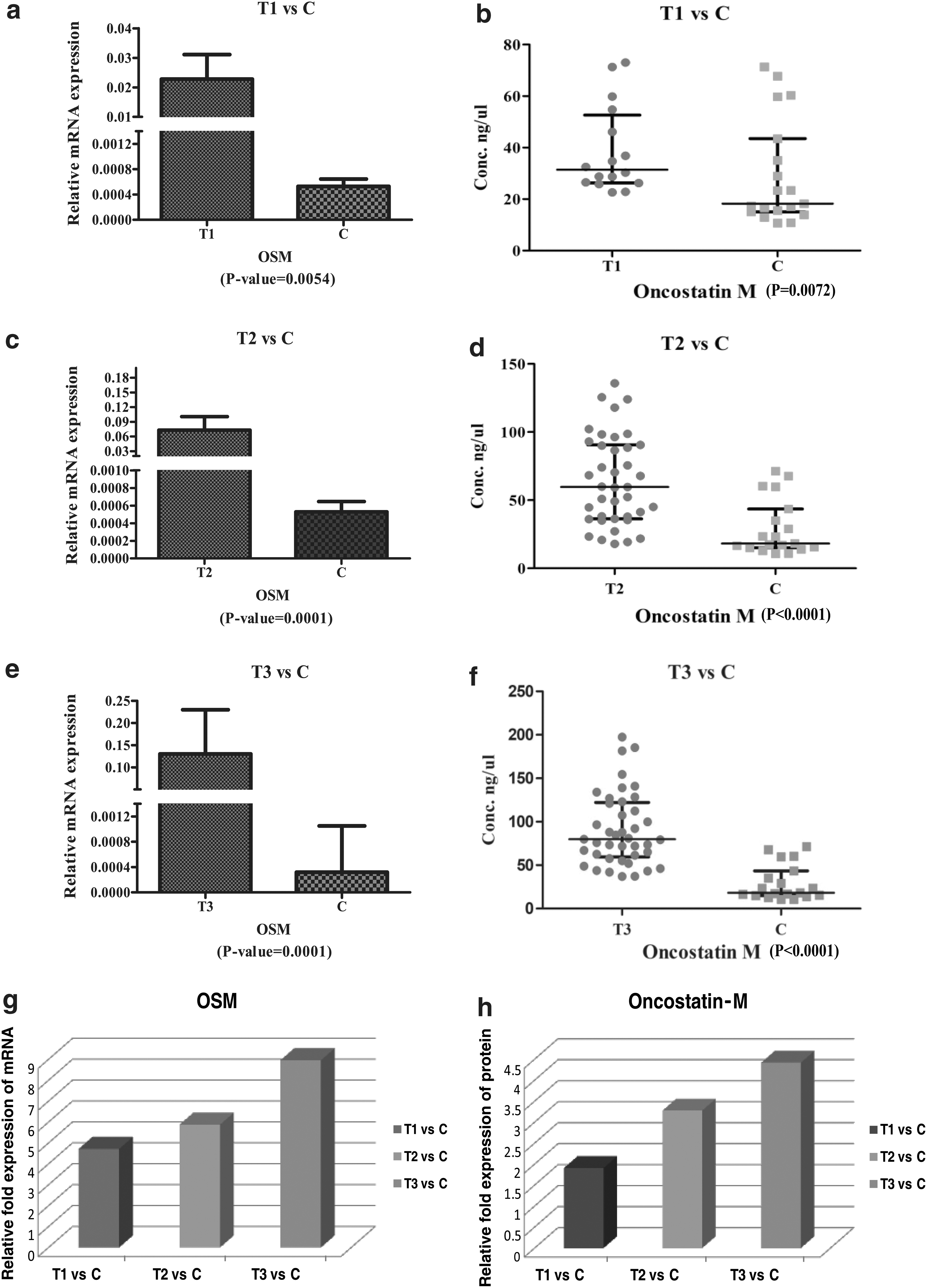
Validation of OSM at both transcript and protein levels. (
Discussion
CV-developed post-aSAH is one of the main causes of neurological deficits. Although CV has been documented for more than half a century, it remains elusive with respect to its pathogenesis, prevention, and treatment. Progress in our understanding of its pathogenesis and systems biology is crucial to reduce the burden of the disease. To the best of our knowledge, this is the first study to compare the transcriptome of aneurysmal tissue samples of aSAH patients with and without CV, and also the first to perform the temporal gene expression analysis.
Various pharmacological and medical interventions have been attempted to overturn the effect of CV, which include, for example, oral nimodipine, statins, lumber drainage of cerebrospinal fluid (CSF), hypertension, hypervolemia, and hemodilution (HHH) therapy, and intra-arterial infusion of vasodilator (nimodipine, among others) (Chan et al., 2021). However, none of these has been established to appreciably modify the overall outcome. It is intriguing that not every aSAH patient develops CV despite similar clinical features such as bleeds, location and size of the aneurysm, clinical characteristics, family history, and comorbidities. Even patients with a good-grade aSAH develop CV, which leads to poor outcome. Despite the various technological advancements in medical science, our understanding of the molecular mechanisms underlying the development of vasospasm after aSAH is incomplete. Therefore, we performed the present untargeted gene expression analysis to identify the dysregulated genes associated with the development of CV after aSAH.
In our previous study, we performed the transcriptome analysis of tissue samples of two clinical subsets of the patients with IA, that is, unruptured IA and ruptured IA without vasospasm along with controls and identified candidate genes and signaling pathways that may be involved in the formation and rupture of IA. In continuation to this, we further performed the transcriptome analysis of the third clinical subset of the patients with IA, that is, ruptured IA with vasospasm. We performed the RNA sequencing of the aneurysm tissue samples obtained from the patients having ruptured IA with vasospasm and compared with the transcriptome data of aneurysm tissue samples of the patients with ruptured IA without vasospasm. Furthermore, we also compared each disease group individually with the control group and performed the temporal gene expression analysis. Notably, this is the first study conducted in this manner on the Indian population and is poised to make a substantial contribution to the literature.
Our study identified that GAS1, LMOD1, ATP1A2, ADH1B, SUSD4, ELN, and RSAD2 genes were significantly dysregulated when we compared the transcriptome of tissue samples of ruptured IA with vasospasm with those without vasospasm. All these genes were found to be upregulated, with RSAD2 having the highest expression. RSAD2 codes for Viperin, which is an interferon inducible protein. Viperin plays a crucial role in the innate immune response against a broad range of viral infections and is involved in the activation of various immune cells, including macrophages, dendritic cells, and T cells (Ghosh and Marsh, 2020; Kim et al., 2019).
Studies have revealed that Viperin is involved in the activation of NF-κB and AP-1 in T cells (Qiu et al., 2009). In addition, Viperin modulates cellular metabolism, suggesting its involvement in various physiological processes beyond immune responses (Choi et al., 2022). It is also detected in the endothelium of human atherosclerotic lesions, indicating its potential role in the pathogenesis of vascular diseases (Olofsson et al., 2005). The role of RSAD2 has not been studied in the pathogenesis of IA and vasospasm. The activation of NF-κB and AP-1 by RSAD2 could play an important role in the development of CV.
The AP-1 gene, also known as activator protein-1, is a transcription factor that plays a crucial role in regulating gene expression in response to various extracellular stimuli. When activated, AP-1 promotes the expression of proinflammatory mediators such as cytokines and proteases. The IL-1 or interleukin-1 is one of the proinflammatory cytokines regulated by AP-1 (Kino and Chrousos, 2005). It has been implicated in the pathogenesis of several diseases, including stroke, where it has been shown to exacerbate ischemic injury (Yamasaki et al., 1995), and promote neuronal cell death (Liu and Quan, 2018). IL-1 can also stimulate the expression of endothelin, which is a potent vasoconstrictor and induces CV (Galea et al., 2011).
Furthermore, AP-1 is involved in regulating the expression of MMP9 (Huang et al., 2014; Tombulturk et al., 2019). The role of this matrix metalloprotease has been extensively studied in the pathophysiology of CV (Kurogi et al., 2015; Sharma et al., 2020). NF-κB's role in the pathogenesis of CV has also been extensively studied. Inflammation is a key factor in the development of CV, and NF-κB plays a central role in this process (Liu et al., 2023). It regulates the transcription of proinflammatory genes such as iNOS and MMPs. It is also involved in the transactivation of genes associated with endothelial dysfunction, such as VACM-1 and MCP-1 (Aoki et al., 2007).
There are studies that established the role of these molecules (MMP9, iNOS, and MCP-1) in the development of CV (Kim et al., 2008; Sayama et al., 1999). Therefore, the upregulation of RSAD2 could play an important role in the pathogenesis of CV by regulating these important molecules.
Another important gene that could play a crucial role in the pathogenesis of CV is ATP1A2. In the present study, we observed the upregulation of this gene. It encodes for the α2-subunit of Na+/K+-ATPase, a transmembrane protein. The Na+/K+-ATPase is responsible for intracellular ion homeostasis, which is essential for membrane potential and various cellular processes. It is involved in the apoptosis and proliferation of vascular smooth muscle cells (VSMCs). The inhibition of Na+/K+-ATPase suppresses the apoptosis of VSMCs (Wang et al., 2022). It is observed that apoptosis of serum-derived VSMCs was sharply suppressed when Na+/K+-ATPase was inhibited with ouabain (Na+/K+-ATPase inhibitor). Furthermore, the role of Na+/K+-ATPase in VSMC proliferation was also demonstrated (Wang et al., 2022).
Vasospastic segments of blood vessels are characterized by the proliferation of smooth muscles and myofibroblast as well as cellular remodeling and necrosis (Borel et al., 2003). The proliferation and phenotypic alterations of VSMCs play an important role in CV (Hu et al., 2023).
In addition, Na+/K+-ATPase can form the complex with Src (Tian et al., 2006). On exposure to ouabain, Na+/K+-ATPase interacts with neighboring membrane proteins, initiating an intricate signaling cascade that includes the activation of Src, EGFR, and Ras (Haas et al., 2002), and leads to the Ras/MAPK cascade (Hu et al., 2018). The ouabain/Na+/K+-ATPase/Src complex could lead to the activation of Ras, promoting the mitochondrial production of reactive oxygen species (ROS), leading to secondary damage to mitochondria, loss of function, and eventually autophagy (Tian et al., 2006; Xie et al., 2003).
Moreover, this complex can activate apoptosis signal regulating kinase 1 and Caspase 3, 8, 9, and promote apoptosis through Fas-mediated apoptosis pathway (Hu et al., 2018). Hence, ATP1A2 might be important for the development of vasospasm. In contrast, the role of Na+/K+-ATPase in vasodilation has also been studied. Its activation in VSMCs leads to membrane hyperpolarization, which eventually reduces the intracellular Ca2+ and contributes to vasodilation. However, its pharmacological inhibition or knocking it down leads to the increased contraction of VSMCs by influencing intracellular Ca2+ concentrations through NCX (Zhang et al., 2019).
The ELN gene encodes the elastin protein, a crucial component of the extracellular matrix (ECM), predominantly found in the internal elastic lamina of intracranial arteries. Observations of defects in this lamina among IA patients have sparked speculation regarding the potential involvement of ELN in IA pathogenesis (Paterakis et al., 2017). Several studies have suggested a plausible association between the ELN gene and IA formation (Jeon et al., 2018). The SUSD4 gene encodes a transmembrane protein containing four Sushi domain motifs, primarily expressed in the central nervous system.
Although direct links between SUSD4 and IA formation or complications such as rupture or vasospasm are yet to be established, its role in complement regulation and inflammation implies potential involvement in IA pathogenesis. Notably, dysregulation of the complement system and inflammation are implicated in both IA development and complications (Boulieris et al., 2023). Elevated expression of plasma complement C3a and ICAM-1 has been correlated with the development of vasospasm and poor outcomes (Findlay et al., 2016).
Furthermore, our study identified dysregulation of the LMOD1 gene. LMOD1 is notably abundant in VSMCs. Genetic variations affecting LMOD1 functionality have been linked to predisposition to thoracic aortic aneurysm and dissection. Despite a lack of clear understanding regarding its role in the pathophysiology of IA, GO analysis indicates its association with smooth muscle contraction and phenotypic modulation. Given the significance of these processes in the pathogenesis of IA and vasospasm, further exploration of the involvement of LMOD1 in IA development is warranted.
Similarly, GAS1 encodes the growth arrest-specific protein 1, which plays a role in growth suppression and apoptosis. Given the significance of apoptosis in aneurysm formation and rupture, it is conceivable that GAS1 may play a crucial role in IA pathogenesis through apoptosis regulation. However, further research is needed to elucidate the precise mechanisms by which GAS1 influences IA development and progression. Our study observes the dysregulation of ADH1B gene that encodes a protein that belongs to the alcohol dehydrogenase family. Research indicates that individuals with the ADH1B rs1229984 TC+CC genotype, coupled with alcohol exposure, may have an elevated risk of developing hemorrhagic stroke (Lin et al., 2021). Its direct relevance to IA remains unexplored, although understanding genetic influences on stroke risk factors could shed light on broader cerebrovascular disease mechanisms.
To obtain the holistic view of the pathophysiology and mechanism of disease progression, we conducted temporal gene expression analysis. We identified four upregulated genes exhibiting temporal patterns: TNFSF13B, PLAUR, OSM, and LAMB3. Notably, OSM displayed a more significant expression change from aneurysmal developmental to rupture and subsequent vasospasm. OSM, a cytokine produced by the activated macrophages, interacts with endothelial cells via various receptors in the vascular wall. It has been shown that endothelial cells are the primary targets of OSM, which not only stimulates endothelial cell proliferation and migration but also regulates the expression of IL-6, P-selectin, and plasminogen activator (Nagata et al., 2013). In addition, it also induces the proinflammatory processes in smooth muscle cells (SMCs) by stimulating the expression of IL-6 and cyclooxygenase-2 (Bernard et al., 1999). The endothelial cell proliferation and phenotypic modulation of SMCs are crucial factors in the formation of IA.
The degradation of the ECM is a prominent feature in the progression and rupture of IA. Studies have reported that OSM induces MMP2 and MMP9 expression in cultured SMCs, highlighting its role in ECM degradation (Nagata et al., 2013). The regulation of ECM metabolism in various cells other than SMCs is also reported to be regulated by OSM by inducing the MMPs (Korzus et al., 1997; Richards et al., 1993). Therefore, overexpression of the OSM could be an important factor in the IA rupture. OSM is further known to stimulate the expression of IL-6 and MCP-1.
Various studies in the past have reported the upregulation of IL-6 and MCP-1 after aSAH and suggest their potent role in the development of vasospasm (Kim et al., 2009; Lu et al., 2009; Muroi et al., 2013; Wang et al., 2007). Hence, based upon the observations, it can be stated that OSM regulating the various inflammatory, vascular remodeling, and ECM degradation processes in endothelial and SMCs is involved in the pathogenesis of the disease at all the stages of formation, progression, rupture, and further development of vasospasm.
Study Limitations
This study is subject to several limitations, with the small sample size being the foremost constraint. Nonetheless, addressing this limitation proved to be challenging due to the intricate nature of harvesting the tissue samples during the IA surgery. A significant number of patients suffering from aSAH typically present at the hospital within 2–3 days postictus, a period during which vasospasm does not commonly manifest. Consequently, the occurrence of preoperative vasospasm is infrequent, which contributes to the limited number of eligible patients for inclusion in the study. However, we performed the validation in a comparatively larger cohort of samples. Ideally, paired samples are preferred for the temporal gene expression analysis. However, collecting paired samples is not feasible in the case of IA.
Conclusions
Using high-throughput next-generation sequencing for gene expression analysis, our study identified the dysregulation of genes in the aneurysm tissue samples of the patients having aSAH with vasospasm when compared with those without vasospasm. Considering both biological and statistical significance, we identified RSAD2 and ATP1A2 as potentially vital genes with clinical relevance, likely contributing to the pathophysiology of CV. Furthermore, as we completed the transcriptome analysis of all clinical subsets of the patients with IA to obtain a holistic view of the pathophysiology of the disease, we performed the temporal gene expression analysis and observed that OSM could be the candidate gene playing an important role in the formation and rupture of IA and consequently in the development of vasospasm. Nonetheless, it is imperative to note that further translation research and validation of these findings warrant consideration, ideally in a multicentric large cohort and complementary functional studies.
Taken together, these findings offer new insights on the molecular basis of CV and point to potential molecular leads for translational research and therapeutic/diagnostic innovation.
Footnotes
Acknowledgments
The authors thank the patients and their family members for participation in this study.
Authors' Contributions
Conceptualization: H.B., M.K., A.C., and H.G. Sample and data collection: M.K., T.S., S.C., and R.D. Samples provided by: A.A., C.G., N.S., R.C., P.S., S.D., and A.T. Research facilities: T.G. and A.P. Bioinformatic analysis: K.P. Data interpretation: M.K., T.S., and H.B. Project administration: H.B. Supervision: H.B., A.C., and H.G. Validation: M.K. and T.S. Visualization: M.K., T.S., and K.P. Writing—original draft preparation: M.K. and H.B. Writing—review and editing: M.K., H.B., T.S., A.C., and H.G. All the authors made a significant intellectual contribution, and read and approved the final article.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
The study was financially supported by the PGIMER, Chandigarh [No. 71/2-Edu-16/4484], through intramural research grant scheme. Munish Kumar was funded by the University Grants Commission (UGC) fellowship. Tanavi Sharma was funded by a PGIMER fellowship. Krishna Patel was funded by CSIR, New Delhi.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.