
Abstract
The axolotl (Ambystoma mexicanum) is renowned for its remarkable regenerative capabilities, which are not diminished by the transition from a neotenic to a metamorphic state. This study explored the microbiome dynamics in axolotl limb regeneration by examining the microbial communities present in neotenic and metamorphic axolotls at two critical stages of limb regeneration: pre-amputation and during blastema formation. Utilizing 16S rRNA amplicon sequencing, we investigated the variations in microbiome profiles associated with different developmental and regenerative states. Our findings reveal a distinct separation in the microbiome profiles of neotenic and metamorphic samples, with a clear demarcation in microbial composition at both the phylum and genus levels. In neotenic 0DPA samples, Proteobacteria and Firmicutes were the most abundant, whereas in neotenic 7DPA samples, Proteobacteria and Bacteroidetes dominated. Conversely, metamorphic samples displayed a higher abundance of Firmicutes and Bacteroidetes at 0DPA and Proteobacteria and Firmicutes at 7DPA. Alpha and beta diversity analyses, along with dendrogram construction, demonstrated significant variations within and between the sample groups, suggesting a strong influence of both developmental stage and regenerative state on the microbiome. Notably, Flavobacterium and Undibacterium emerged as distinctive microbial entities in neotenic 7DPA samples, highlighting potential key players in the microbial ecology of regeneration. These findings suggest that the axolotl’s microbiome is dynamically responsive to blastema formation, and they underscore the potential influence of microbial communities on the regeneration process. This study lays the groundwork for future research into the mechanisms by which the microbiome may modulate regenerative capacity.
Introduction
Axolotl has intrigued scientists for centuries because of its extraordinary regenerative capabilities (Sámano et al., 2021). This species, which uniquely retains its larval characteristics into adulthood, has the remarkable ability to regenerate entire limbs, tail, heart, and other organs with perfect fidelity (Coots and Seifert, 2015). Unlike wound healing in mammals, which typically results in scar tissue formation, axolotl regeneration is a meticulous process of tissue, bone, and organ reconstruction without any residual scarring, thus maintaining full functionality of the regenerated structure (Endo et al., 2004).
The regenerative excellence of the axolotl is evident by the formation of the blastema following limb amputation. This regeneration-specific tissue, characterized by the accumulation of stem and progenitor cells capable of growth and differentiation, is skin to a biological scaffold where the regrowth of the limb is outlined (Hyun et al., 2012). This structure is not merely a cluster of cells but a dynamic, organized tissue with a defined pattern of gene expression that guides the regeneration process (Bassat and Tanaka, 2021). The blastema’s significance extends beyond its regenerative capacity; it serves as a model for understanding cellular dedifferentiation and reprogramming, processes that are fundamental to the development of regenerative medicine and tissue engineering (Tsai et al., 2019).
The complex molecular mechanisms governing the process of axolotl limb regeneration have become a prominent focus of comprehensive investigation. Research endeavors concentrating on the phenomenon of axolotl blastema have provided valuable insights into the coordination of gene expression, the functions of various signaling molecules in facilitating cell-to-cell communication, as well as the dynamic interplay between cellular and extracellular constituents throughout the regeneration process (McCusker et al., 2015). These investigations are driving the field closer to uncovering conserved genetic networks and signaling pathways that have the potential to be leveraged for the purpose of inducing regenerative responses in species that do not naturally possess this capability, such as the human species (Alibardi, 2023; Seifert and Muneoka, 2018).
Regeneration is marked by a dynamic alteration in the localized and systemic biological environment, encompassing modifications in the microbiome (Díaz-Díaz et al., 2022). The concept of the microbiome, the collective genome of all microorganisms residing in or on an organism, has revolutionized our understanding of host–microbe interactions and their impact on host physiology (Kostic et al., 2013). The microbiome is currently acknowledged as a critical participant in diverse biological processes, spanning from nutrient assimilation and metabolism to the modulation of immune responses (Wang et al., 2017) The microbiome’s role in regeneration is a nascent field of study, but existing research suggests that microbial communities could influence regenerative outcomes by modulating host immune responses, providing biochemical signals for cell proliferation and differentiation, and impacting the local tissue microenvironment (Wu and Wu, 2012). Changes in the microbiome throughout the regeneration process could hence yield novel insights into enhancing regenerative responses, with these interactions potentially playing a crucial role in the successful renewal of tissues (Shavandi et al., 2020).
In this context, we employed 16S ribosomal RNA (rRNA) amplicon sequencing to delineate the microbiome composition associated with the axolotl’s limbs during two critical stages: the intact limb before amputation (0 days post-amputation, DPA) and the early phase of regenerating limb characterized by the formation of the blastema (7DPA). Our comprehensive analysis encompasses a comparison between the microbiomes of neotenic and metamorphic axolotls, thereby elucidating the influence of developmental status on microbial composition during limb regeneration. The present study aimed to deepen our understanding of the microbiome’s role in the axolotl’s regenerative process and to unravel the complex interplay between host and microbial communities during tissue renewal, with the hope of developing microbiome-based strategies in regenerative therapies and advancing the field of regenerative biology.
Materials and Methods
Research ethics
The research protocol involving animals and their maintenance conditions received approval from the Istanbul Medipol University (IMU) Ethics Committee, granted under the authorization number 10.1353/pbm.2019.0046.
Animal husbandry and induction to metamorphosis
This research incorporated 36 adult axolotls (1 year old, 12–15 cm in length), selected at random from a cohort of siblings. The progenitors of the original axolotl colony were acquired from Kentucky, USA, and have been propagated and cared for at the IMU animal facility in accordance with established husbandry protocols. Axolotls were housed individually in aquaria at a controlled temperature of 18 ± 2°C in Holtfreter’s solution. The university’s animal facility maintained a batch-flow aquarium system equipped with UV sterilization and filtration to mitigate infection risks. All axolotls were exposed to identical aquatic conditions and fed a uniform diet. The daily diet consisted of JBL Novo LotlM (Neuhofen, Germany), and no antibiotic interventions were implemented throughout the experiment.
From the total sample size, 18 axolotls were designated for metamorphosis following methodologies outlined in our previous study (Demircan et al., 2018) via L-thyroxine (Sigma-Aldrich, St. Louis, MO, USA, Cat. No. T2376) administration. In brief, an aqueous solution of L-thyroxine was dissolved in Holtfreter’s solution to achieve a final concentration of 50 nM and was applied to the specimens housed individually for the duration of the experiment. The medium containing T4 was renewed every third day. The subjects were observed for morphological changes such as reduced mass, fin regression, and gill shrinkage, which typically manifested within 3 weeks post T4 administration. Hormonal treatment was extended for an additional 3 weeks to yield fully metamorphosed axolotls. After metamorphosis and before limb removal and sample collection, metamorphosed animals were allowed a 1-month period without T4 treatment to acclimatize to terrestrial living conditions.
Experimental design
Limb amputation was conducted under anesthesia induced by 0.1% tricaine methane sulfonate (Cat. No. E10521 or MS-222, Sigma-Aldrich), severing the right forelimb at the mid-zeugopod level. Post-amputation, the subjects were allocated into two distinct groups to represent the intact phase and the early blastema stage for both neotenic and metamorphic classifications. For each experimental group, three biological replicates (R1, R2, and R3) were formed to validate the reproducibility of the results. To reduce individual variability, tissue samples from three specimens were pooled for each biological replicate. Tissue samples were preserved instantaneously in liquid nitrogen post-collection and stored at −80°C till genomic DNA extraction. Tissue surrounding the amputation site was harvested from 0DPAspecimens, whereas the nascent blastema and an additional 0.5 mm of adjacent tissue were excised from 7DPAspecimens as previously described (Demircan et al., 2019).
DNA extraction and 16S amplicon sequencing
Genomic DNA was extracted utilizing the DNeasy Blood and Tissue kit (Qiagen, Germany), according to the manufacturer’s instructions. The extracted DNA was quantified through the Qubit system, and its quality was verified by electrophoresis in a 1% Agarose gel. The extracted DNA served as the template for amplifying the V3–V4 region of the 16S rRNA gene using specific primers, 337F (5′-GACTCCTACGGGAGGCWGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′), which had Nextera sequencing adaptor sequences appended to their ends. The polymerase chain reaction (PCR) mixtures, with a volume of 25 µL, comprised 12.5 ng of DNA and 2× KAPA HiFi HotStart Ready Mix. The PCR thermal cycling involved an initial denaturation at 95°C for 3 min, followed by 25 cycles of denaturation at 95°C for 30 sec, annealing at 55°C for 30 sec, and extension at 72°C for 30 sec, with a concluding extension at 72°C for 5 min.
The PCR products underwent purification using the Agencourt AMPure XP system (Beckman Coulter, Cat. No. A63881, USA) and were subjected to a second round of PCR for eight cycles to incorporate sample-specific barcodes. The amplicons were purified again post-barcoding. Libraries, standardized to equimolar concentrations, were sequenced using an Illumina MiSeq platform with a 500-cycle MiSeq Reagent Kit v2. The average sequence length was approximately 231 base pairs. As a control, aqua samples of neotenic and metamorphic samples were sequenced and analyzed.
Raw data processing and downstream analysis of 16S rRNA sequencing
Raw sequence data processing was carried out following our previously established pipeline (Demircan et al., 2018; Demircan et al., 2019) commenced with the Illumina BaseSpace platform for demultiplexing and the removal of adapters and barcodes. Primer sequences were then trimmed from the raw 16S rRNA paired-end sequences using cutadapt (version 1.13). The processed sequences underwent quality checks using FastQC, part of the Nephele Pre-processing QC pipeline, with any reads scoring a Phred value below 30 being discarded. The remaining high-quality sequences were analyzed with QIIME2 (Bolyen et al., 2019) against the SILVA v.132 database using a closed operational taxonomic unit (OTU) strategy at 97% sequence identity. The resulting OTU table facilitated a range of downstream analyses including data scrutiny, normalization, abundance visualization, and diversity assessments. These analyses were conducted using the MicrobiomeAnalyst web tool (Lu et al., 2023), particularly the Marker Data Profiling module. Initial filtering removed low-abundance and low-prevalence features, followed by the exclusion of low-variance features using an interquantile range threshold of 5%. Data normalization was achieved through total sum scaling, without rarefying or transforming the data. OTU relative abundances were visualized, with phylum and family-level distributions presented in a stacked barplot format.
Alpha diversity was evaluated using various indices to measure microbial richness and evenness within the samples. The Kruskal–Wallis test, followed by post-hoc Dunn’s test, was used to detect alpha diversity variances among the samples. Beta diversity analysis, including Bray–Curtis and Shannon metrics, and statistical tests, such as the permutational multivariate analysis of variance (PERMANOVA), permutational multivariate analysis of dispersion (PERMDISP), and analysis of similarity (ANOSIM), assessed sample dissimilarities. Visual clustering was depicted through principal coordinate analysis (PCoA) and non-metric multidimensional scaling (NMDS) plots. A dendrogram was constructed using the Bray–Curtis as dissimilarity metric, and Ward’s method as the clustering algorithm. Linear discriminant analysis effect size (LEfSe) analysis identified potential indicator species and biomarkers, applying a stringent linear discriminant analysis (LDA) score and false discovery rate (FDR)-adjusted p-value thresholds. EdgeR was used to discern differences between groups, applying a log2 fold change, p-value, and FDR criteria.
Results
In this comprehensive microbiome analysis, a total of 18 samples (12 axolotl and 6 aqua samples) underwent sequencing and subsequent examination to characterize the microbial communities present during different stages of limb regeneration. To ensure the reliability of the results, the analysis was conducted in duplicate, with or without control samples from the aquatic environment for both neotenic and metamorphic axolotls, providing a comparative context to the biological samples. The assortment of samples comprised three neotenic aquarium water controls, three metamorphic aquarium water controls, three from neotenic axolotls at 0DPA, three from neotenic axolotls at 7DPA, three from metamorphic axolotls at 0DPA, and three from metamorphic axolotls at 7DPA. The sequencing efforts yielded a range of reads per sample, with the lowest being 25070 and the highest reaching 65504, reflecting a substantial amount of data for analysis (Supplementary Fig. S1).
Before the commencement of the analysis, the data were subjected to a rigorous filtration process aimed at augmenting the dependability of the ensuing interpretations. This particular procedure involved the elimination of characteristics that exhibited both a scarcity in occurrence, with a minimum count threshold of one, and a scarcity in prevalence, manifesting in fewer than 10% of the specimens. In addition, characteristics demonstrating limited variability were also dismissed, utilizing an interquantile range cutoff established at 5%. These pivotal stages in data refinement played a crucial role in honing the dataset to 7,038 features, consequently refining the focus toward the most pertinent microbial entities from a biological perspective. Standardization of the dataset was executed through the utilization of the Total Sum Scaling technique, a determination based on the preservation of the original data framework to prevent data rarefaction or alteration, thus upholding the integrity and initial intricacy of the dataset.
Species richness and diversity within the axolotl limb microbiome were quantitatively assessed using alpha-diversity metrics, including Chao1, Shannon, Simpson, and observed species indices. Among the metrics evaluated, samples from neotenic axolotls at 7DPA consistently presented with the lowest diversity and richness values across all taxonomic levels assessed (Fig. 1A). Pairwise comparisons between the experimental groups were executed to ascertain the presence of statistically significant differences in microbial diversity (Fig. 1A). The observed species index indicated a significant disparity between neotenic samples at 0DPA and those at 7DPA, with a p-value of 0.046. Similarly, a notable difference was observed in the Shannon index between metamorphic samples at 0DPA and 7DPA (p = 0.0369). Furthermore, the Simpson index underscored a significant divergence for the same metamorphic comparisons (p = 0.0166) (Fig. 1A). These findings substantiate the presence of statistically significant alterations in the microbiome's composition and structure between the distinct stages of limb regeneration, highlighting the dynamic nature of microbial communities in response to the regenerative process.
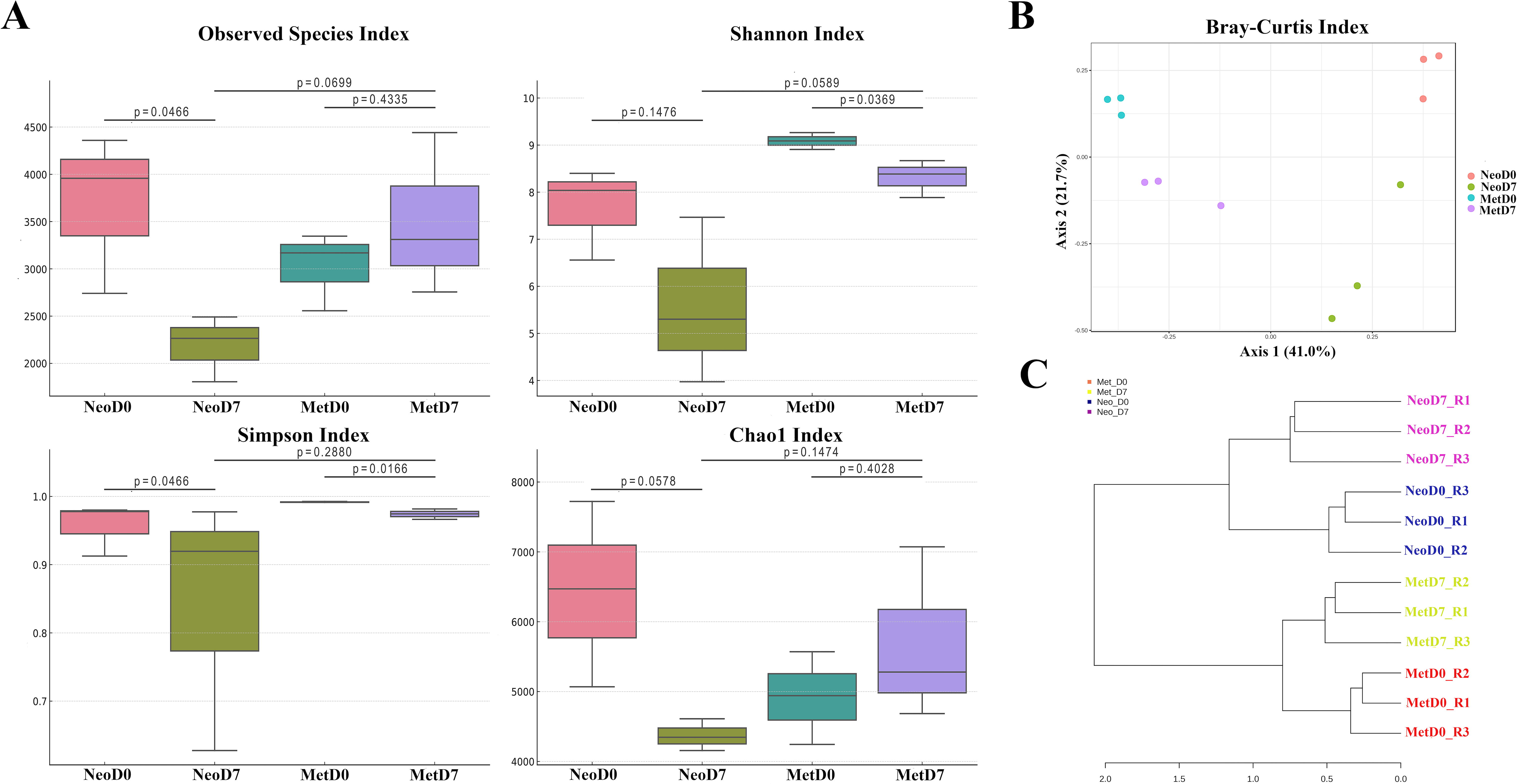
Microbial diversity comparison in axolotl limb samples.
The beta diversity, which reflects the compositional heterogeneity between microbial communities, was quantified using Bray–Curtis and Jaccard distance metrics to evaluate the distinctions among the bacterial populations in the axolotl samples (Fig. 1B). The resulting analyses elucidated a pronounced differentiation across the groups. PERMANOVA reinforced these observations, yielding statistically significant differences for both utilized metrics. Specifically, the Bray–Curtis index produced a Pseudo-F value of 5.0472, an R-squared value of 0.6543, and a p-value (Monte Carlo) of 0.001, demonstrating substantial dissimilarity in community composition (Table 1). Similarly, the Jaccard index indicated significant beta diversity with a Pseudo-F value of 3.3036, an R-squared value of 0.55334, and a Monte Carlo p-value of 0.001. Further validation of these results came from the ANOSIM, which provided support for the beta diversity findings, yielding an R value of 0.82716 and a p-value of <0.001 for the Bray–Curtis index and an R value of 0.74815 with a p-value of <0.001 for the Jaccard index. These high R values from ANOSIM indicate that the similarities within groups are much smaller than the dissimilarities between groups. The harmony of these significant results was visually substantiated through both PCoA and NMDS. These visualizations corroborated the consistency of the beta diversity findings and marked differences in microbial community structure among the axolotl limb samples at different stages of regeneration.
Comparative Analysis of Beta Diversity Indices and Statistical Methods in Axolotl Samples
A comparative analysis of beta diversity indices applied to axolotl samples, evaluating the significance of community differences using various statistical approaches and the resulting p-values. p < 0.05 was considered statistically significant.
ANOSIM, analysis of similarity; PERMANOVA, permutational multivariate analysis of variance.
Moreover, the dendrogram, generated based on feature-level clustering, depicted a clear segregation of neotenic and metamorphic samples, signifying distinct microbiome profiles associated with each developmental stage (Fig. 1C). In addition, within these developmental clusters, samples from the 0DPA were distinctly separated from the 7DPA samples, underscoring the dynamic nature of the microbiome during the course of regeneration (Fig. 1C).
The taxonomic composition of the axolotl limb microbiome displayed a dynamic shift from 0DPA to 7DPA, both in neotenic and in metamorphic samples (Fig. 2). This alteration was evident at the phylum level where, in neotenic 0DPA samples, Proteobacteria dominated with 52.5%, followed by Firmicutes at 18.9%, Actinobacteriota at 11.6%, and Bacteroidetes at 9.9%. In contrast, neotenic 7DPA samples revealed a different distribution, with Proteobacteria at 43.3%, Bacteroidetes surging to 34.4%, Firmicutes at 11.9%, and Actinobacteriota reduced to 5.0% (Fig. 2A). The metamorphic samples followed a similar trend of fluctuation. Initially, metamorphic 0DPA samples presented Firmicutes as the most abundant phylum at 53.2%, Bacteroidetes at 19.7%, Proteobacteria at 12.9%, and Verrucomicrobiota at 7.3%. However, by the metamorphic 7DPA stage, Proteobacteria rose to 33.8%, whereas Firmicutes decreased to 29.3%, and Bacteroidetes increased to 25.3%, with Actinobacteriota now accounting for just 2.0% (Fig. 2A). At the family level, the neotenic 0DPA samples predominantly harbored Comamonadaceae (30.5%), Streptococcaceae (9.4%), Propionibacteriaceae (7.0%), and Moraxellaceae (5.0%). Upon reaching the neotenic 7DPA stage, there was a noteworthy dominance of Flavobacteriaceae (40.0%), a substantial presence of Comamonadaceae (18.4%), and lesser abundances of Aeromonadaceae (6.8%) and Oxalobacteraceae (6.5%) (Fig. 2B).
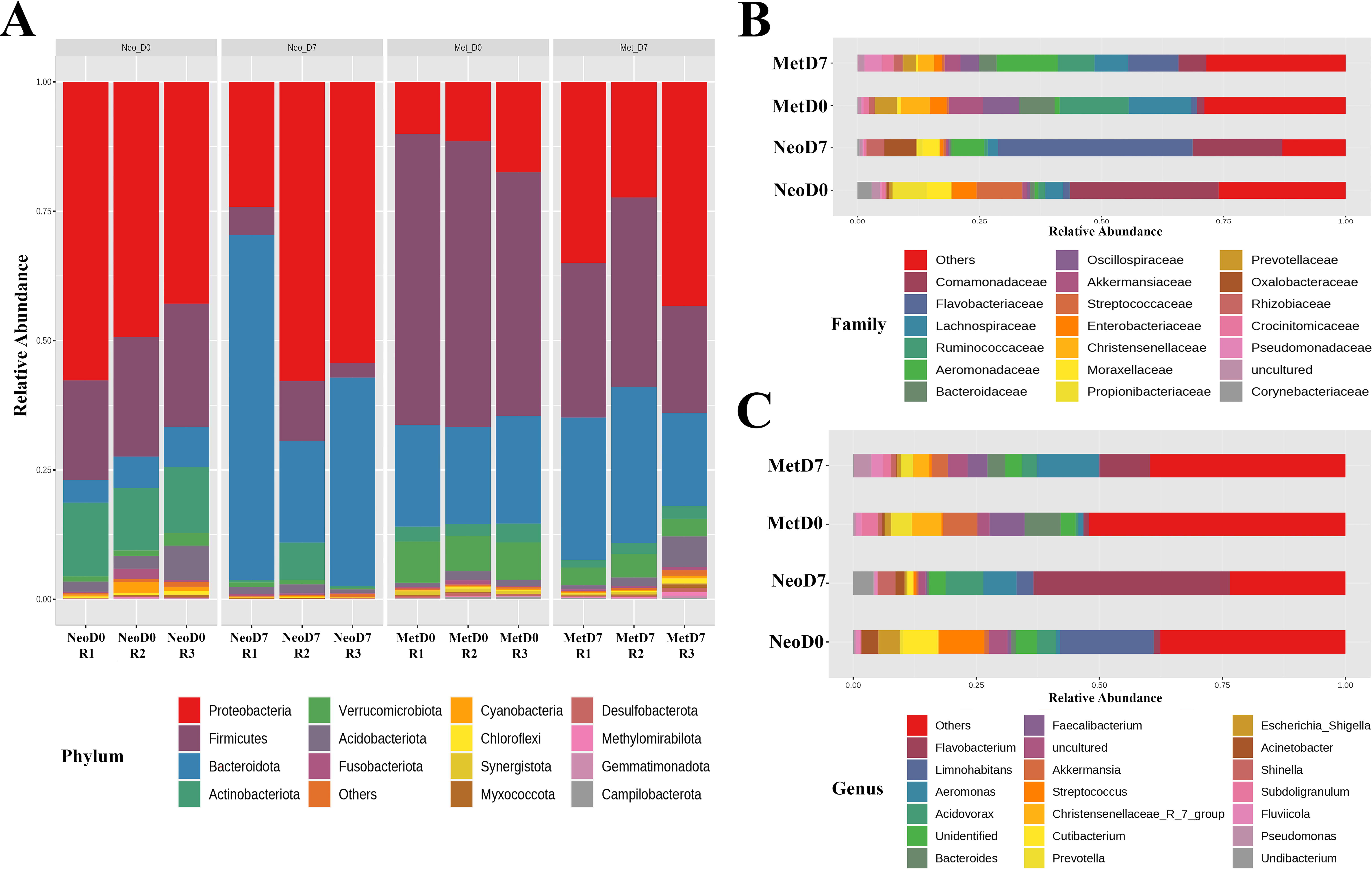
Comparative microbial community analysis in intact and regenerating axolotl limb samples. An in-depth comparison of microbial community structures of axolotl samples, specifically emphasizing the taxonomic units with the highest abundance.
In the metamorphic 0DPA samples, the leading families were Ruminococcacaea (14.1%), Lachnospiraceae (12.8%), Oscillospiarceae (7.4%), and Bacteroidaceae (7.3%). By the metamorphic 7DPA, Aeromonadaceae emerged as the most prevalent (12.5%), with Flavobacteriaceae (10.2%), Ruminococcacaea (7.4%), and Lachnospiraceae (6.9%) also featuring prominently (Fig. 2B). In the neo 0DPA samples, Limnohabitans was the predominant bacterial genus, constituting 19% of the population, with Streptococcus following at 9.3% (Fig. 2C). At the 7DPA stage within neotenic axolotls, Flavobacterium surged to become the most abundant at 40%, and Acidovorax was next, representing 7.6% of the bacterial community. Metamorphic 0DPA samples displayed a different profile, with Bacteroides and Faecalibacterium being the most prevalent, at 7.3% and 7.1%, respectively (Fig. 2C). In metamorphic 7DPA samples, Aeromonas, at 12.6%, and Flavobacterium, at 10%, were identified as the dominant genera, marking a shift in microbial dominance as the regeneration process progresses.
Core microbiome analysis and biomarker identification
The core microbiome represents the microbial taxa consistently found across all samples which might be an indicator of potentially stable and essential microbial component within a given ecological niche or biological context. Upon assessing the core microbiome at the family level (Fig. 3A) for neotenic 0DPA samples, the following taxa were ubiquitously present across all replicates: Streptococcaceae, Staphylococcaceae, Ruminococcaceae, Propionibacteriaceae, Moraxellaceae, Lactobacillaceae, Lachnospiraceae, Enterobacteriaceae, Corynebacteriaceae, and Comamonadaceae. Similarly, for neotenic 7DPA samples, Oxalobacteraceae, Lachnospiraceae, Flavobacteriaceae, Comamonadaceae, and Aeromonadaceae were detected in every replicate. Notably, a more stable core microbiome was observed in the metamorphic 0DPA and 7DPA samples in comparison with the neotenic counterparts. Metamorphic 0DPA samples exhibited a core microbiome consisting of Veillonellaceae, Tannerellaceae, Ruminococcaceae, Rikenellaceae, Prevotellaceae, Oscillospiraceae, Lachnospiraceae, Eubacterium_coprostanoligenes_group, Enterobacteriaceae, Comamonadaceae, Clostridia_UCG_014, Christensenellaceae, Bacteroidaceae, and Akkermansiaceae. Finally, the core microbiome for metamorphic 7DPA samples included Ruminococcaceae, Rhizobiaceae, Pseudomonadaceae, Prevotellaceae, Oscillospiraceae, Lachnospiraceae, Flavobacteriaceae, Enterobacteriaceae, Comamonadaceae, Christensenellaceae, Bacteroidaceae, Akkermansiaceae, and Aeromonadaceae. These findings pinpointed a substantial similarity between metamorphic and a noteworthy difference between neotenic samples in terms of core microbiome.
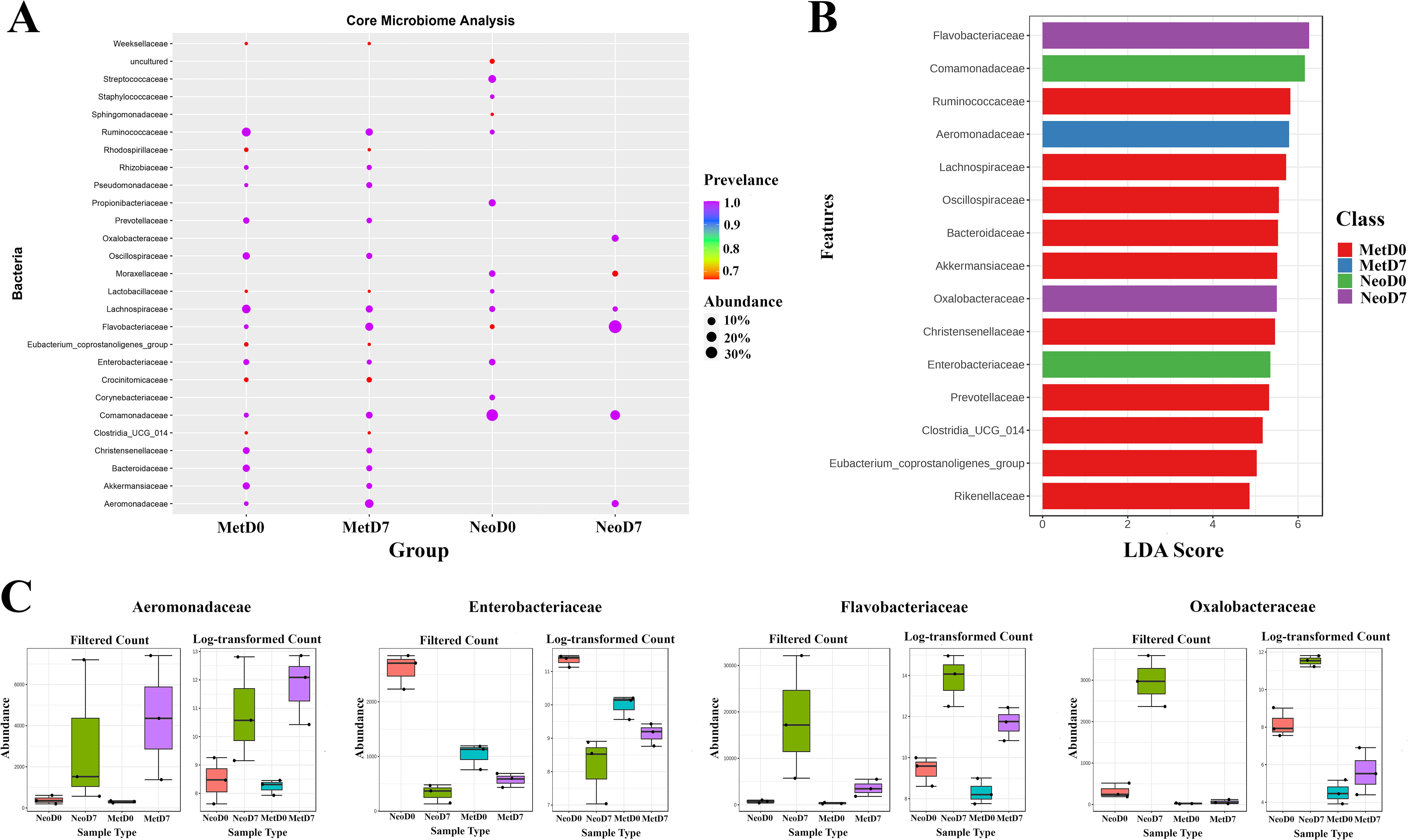
Familial commonality and diversity within axolotl microbial communities.
LEfSe analysis was then used to identify taxa that are differentially abundant between groups and can serve as potential biomarkers by integrating linear discriminant analysis with statistical tests to determine the features most likely to explain differences between classes (Fig. 3B). According to LEfSe results, Comamonadaceae and Enterobacteriaceae were identified as potential biomarkers for Neotenic 0DPA, Flavobacteriaceae and Oxalobacteraceae for Neotenic 7DPA, Lachnospiraceae and Ruminococcaceae for Metamorphic 0DPA, and Aeromonadaceae for Metamorphic 7DPA.
As a separate analysis, EdgeR, a bioinformatic tool used to identify differentially abundant features within the taxa, was applied. EdgeR conducts differential abundance analyses using a model based on the negative binomial distribution, ideal for count-based sequencing data such as that generated from 16S rRNA gene sequencing. Utilizing EdgeR, 85 taxa were flagged as differentially abundant across the groups. Notably, four of these taxa (Enterobacteriaceae, Oxalobacteraceae, Aeromonadaceae, and Flavobacteriaceae) overlapped with those identified by LEfSe, underscoring their significance. The intersecting taxa, identified as crucial microbial components in Figure 3C, offer a concentrated insight into the microbial interactions occurring during the process of axolotl limb regeneration.
Control aqua samples and comparative microbial diversity analysis
Aqua samples from both neotenic and metamorphic aquariums were incorporated as controls to establish a baseline for the microbiome composition within the aquatic environment that axolotls inhabit. Alpha diversity analyses incorporating these control samples indicated a lower level of richness and diversity in the aqua environment compared to the axolotl limb samples (Fig. 4A). This was reflected by the lower scores across various metrics, highlighting a reduced microbial complexity in the aqua samples. When exploring the statistical significance of these differences, the observed species and Chao1 indices suggested a distinct microbial population between neotenic and metamorphic aqua samples with FDR-corrected p-values <0.05, whereas for the Shannon and Simpson indices, the differences were not statistically significant (FDR-corrected p > 0.05; Table 2).

Microbial composition and diversity analysis in axolotl and aqua samples.
Alpha Diversity Indices and Pairwise Comparisons with Statistical Significance in Axolotl and Aqua Samples
The outcomes of pair-wise comparisons using various alpha diversity indices, such as Chao1, Observed Species, Shannon, and Simpson. The table includes FDR-corrected p-values to assess the statistical significance of diversity differences between axolotl limb samples at different stages of regeneration and their corresponding aquatic environments. p < 0.05 was considered statistically significant.
FDR, false discovery rate.
Further pair-wise comparisons emphasized that the neotenic aqua samples differed from the neotenic 0DPA samples, as indicated by observed species, Chao1, and Shannon indices (FDR-corrected p < 0.05), but such a difference was not evident for the Simpson index (FDR-corrected p > 0.05). However, no significant differences were detected between the neotenic aqua and neotenic 7DPA samples across all metrics (FDR-corrected p > 0.05). The metamorphic comparisons revealed a significant distinction between aqua and 0DPA samples, as well as between aqua and 7DPA samples, across observed species, Chao1, and Shannon indices (FDR-corrected p < 0.05). The Simpson index, however, did not show significant differences for the comparisons between metamorphic aqua and both metamorphic 0DPA and 7DPA samples (FDR-corrected p > 0.05).
Beta diversity analyses underscored these findings, with PERMANOVA testing using both Bray–Curtis and Jaccard distance metrics yielding a p-value of 0.001, indicating a highly significant difference among groups (Table 3, Fig. 4B). ANOSIM supported this with p-values <0.001 for both distance metrics. However, the PERMDISP showed no significant dispersion among the groups (p > 0.05). In contrast, the mixed-effects randomized Kenward–Roger approximation test (MIRKAT) identified significant differences with p-values <0.05, adding an additional layer of understanding to the spatial distribution of microbial communities in the axolotl aqua environment compared to the regenerating limb tissue.
Assessment of Beta Diversity and Statistical Correlations in Axolotl and Aqua Samples
A detailed comparison of beta diversity indices and their associated statistical evaluations for axolotl limb and aqua samples, highlighting the degree of difference and significance in microbial community composition. The tabulated data include the results from PERMANOVA, ANOSIM, PERMDISP, and MIRKAT analyses, revealing the F-values, R-values, and corresponding p-values for Bray–Curtis and Jaccard indices. p < 0.05 was considered statistically significant.
MIRKAT, mixed-effects randomized Kenward–Roger approximation test; PERMDISP, permutational multivariate analysis of dispersion.
At the phylum level, the microbiome profiles of these aqua samples were similar, with Bacteroidetes and Proteobacteria being predominant (Fig. 4C). Neotenic aqua samples comprised 59.8% Bacteroidetes and 36.1% Proteobacteria, closely mirrored by the metamorphic aqua samples, which were made up of 70.2% Bacteroidetes and 29.5% Proteobacteria. At the family level, the main constituents in neotenic aqua samples were Flavobacteriaceae (35.9%), Comamonadaceae (18.8%), Spirosomaceae (18.5%), and Neisseriaceae (6.6%). In metamorphic aqua samples, Flavobacteriaceae was even more dominant at 48.9%, followed by Aeromonadaceae (20.0%), Spirosomaceae (17.1%), and Sphingobacteriaceae (3.5%).
Concluding our microbial profiling, LEfSe analysis encompassed all groups, including the aqueous control samples (Fig. 4D). At the family taxonomic level, Aeromonadaceae was distinguished as the indicator family for the metamorphic aquatic environment. Conversely, Microbacteriaceae was identified as the biomarker for the neotenic aquatic setting. Within the limb samples, Comamonadaceae achieved the highest LDA score for neotenic 0DPA samples, indicative of its significant association with this intact limb stage. For neotenic 7DPA samples, Oxalobacteraceae stood out as a significantly distinct taxonomic entity. Meanwhile, Ruminococcaceae was pinpointed as the defining biomarker for metamorphic 0DPA samples, exhibiting the largest LDA score among its group, signifying its potential role in the early stages of metamorphic regeneration.
Discussion
The axolotl (Ambystoma mexicanum), known for its extraordinary regenerative capabilities, serves as a fruitful biological model for unraveling the complexities of tissue regeneration. Despite its significance, a comparative analysis of the microbiota between neotenic and metamorphic stages during limb regeneration in this species had remained uncharted territory until now. The primary objective of our study was to delineate and contrast the bacterial microbiota associated with neotenic and metamorphic axolotls, specifically examining the intact limb and the initial phase of regeneration.
In this investigation, we inquired the microbial landscapes of neotenic and metamorphic axolotls to determine if their limb samples differ microbiologically. It is well documented that the axolotl’s ability to regenerate its limbs is significantly diminished following metamorphosis (Demircan et al., 2018; Demircan et al., 2019; Monaghan et al., 2014). Our research sought to elucidate the microbial constituents associated with both neotenic and metamorphic axolotls and to establish whether there exists a correlation between the decline in regenerative capacity post-metamorphosis and the transformation within the microbial community. By controlling environmental variables—such as the composition of the aquarium solutions, ambient temperature, and dietary provisions—we endeavored to prevent the possible contribution from environmental conditions and unveil the potential linkage between the altered microbiota and the diminished regenerative capacity of metamorphic axolotls.
Our investigation underscores a distinct variation in the microbiomes of neotenic and metamorphic axolotl limbs, echoing the findings of prior research. A precedent study by Demircan and his colleagues, which examined the gut, stomach, feces, and skin microbiomes, showed that metamorphosis significantly alters the microbial composition in axolotls, suggesting that such restructuring is a recurrent theme irrespective of the tissue type examined (Demircan et al., 2018). Similarly, research by Martínez-Ugalde et al. observed that the skin bacterial communities of A. altamirani, another axolotl species, are heavily influenced by the metamorphic state of the organism, as well as by seasonal shifts in environmental factors like temperature and water chemistry (Martínez-Ugalde et al., 2022). In a comparable manner, research conducted on A. mavortium—a different member of the Ambystoma genus—observed a significant decrease in the ratio of specific bacterial taxa following metamorphosis (Goodwin et al., 2022). Expanding beyond axolotls, Hartmann et al. illustrated that the cutaneous microbiome of Notophthalmus perstriatus, a new species, exhibited notable differences between paedomorphic and post-metamorphic phases (Hartmann et al., 2023). Similarly, distinct gut microbiome communities were identified across various life stages of the eastern newt (N. viridescens) as delineated by a recent study (Fontaine et al., 2021). The findings of this study are congruent with these studies, reinforcing the hypothesis that metamorphosis acts as an essential determinant shaping the composition of host-associated microbial communities.
Furthermore, our study marks a significant advancement by documenting pronounced alterations in the microbiome’s structure and composition, particularly evident in the neotenic blastema. This contribution is of notable significance within the realm of axolotl limb regeneration, an area that has previously received limited exploration. Building upon a previous report (Demircan et al., 2019), which analyzed 16S rRNA amplicon datasets from axolotl limbs at various days, our study extends the understanding of microbial dynamics during the regenerative process. Although earlier research was only confined to neotenic axolotls, it revealed microbiota shifts during regeneration that resonate with our findings. The phyla Bacteroidetes, Firmicutes, Proteobacteria, Actinobacteria, and Verrucomicrobia were predominant in both intact and regenerating limbs, highlighting a consistency in microbial compositions that our study corroborates.
A vital observation in our research is the distinct microbial communities in axolotl tissues compared to the control aquatic environments. This suggests a selective colonization process by microorganisms in axolotl limb tissues. Specifically, the elevated levels of Bacteroidetes in the 7DPA samples of both neotenic and metamorphic axolotls seem influenced by their aquatic surroundings, where Bacteroidetes dominate. The marked presence of Flavobacteriaceae in the 7DPA samples, especially in the neotenic variant, further points to environmental influence on the microbial composition of regenerating limbs. The concurrent rise of Comamonadaceae in neotenic 7DPA samples and Aeromonadaceae in metamorphic 7DPA samples implies a correlation between dominant bacteria in regenerating tissues and those in aquatic environments.
The roles of Comamonadaceae and Aeromonadaceae in the context of regeneration, however, remain elusive because of the scarcity of prior studies. Comamonadaceae, primarily environmental bacteria, have diverse habitats and roles, ranging from pathogens to symbionts (Jara et al., 2018). Aeromonadaceae, predominantly aquatic, are known for their pathogenic potential in both aquatic animals and humans (Huys, 2014), challenging us to discern their potential involvement in axolotl regeneration.
Significantly, members of Flavobacteriaceae, a family widely distributed in both terrestrial and aquatic ecosystems, have been linked to metabolic diversity and pathogenicity (Gavriilidou et al., 2020; Langenheder and Lindström, 2019). Prior investigations have elucidated that the intestinal microbiome of sea cucumbers undergoes considerable alterations in both community structure and functional attributes during intestinal regeneration, with a noteworthy increase in the prevalence of the Flavobacteriaceae family (Langenheder and Lindström, 2019; Weigel, 2020; Zhang et al., 2020). The elevated presence in the intestinal microbiota of sea cucumbers during regeneration, as indicated in previous studies, suggests a potential role as keystone taxa (Zhang et al., 2019; Zhang et al., 2020). This is further supported by the varied abundance of Flavobacteriaceae and Rhodobacteriaceae in sea cucumbers with distinct regeneration rates, which hints at a possible functional role in the regenerative process. The enzymatic capabilities of Flavobacteriaceae, particularly in degrading various biopolymers, might confer a regenerative advantage (Zhang et al., 2020), a hypothesis that could extend to the axolotl limb regeneration context.
The nuanced differences in opportunistic bacteria profiles between neotenic and metamorphic 7DPA axolotl samples, particularly concerning Flavobacteriaceae ratios, present a significant finding in our dataset. A notable aspect of this observation is the differential colonization of Flavobacteriaceae in metamorphic blastema tissues despite the similar microbial compositions of the aquariums housing both neotenic and metamorphic axolotls. This disparity in colonization rates could be attributed to the altered tissue organization that accompanies amphibian metamorphosis (Demircan et al., 2016; Zhu et al., 2020; Wiechmann and Wirsig-Wiechmann, 2003). Given the direct relationship between host–microbial interactions and tissue structure (Casadevall and Pirofski, 2000; Hooper et al., 2012), this might elucidate the variance in Flavobacteriaceae levels observed in neotenic and metamorphic blastema samples.
Crucially, the maturation of the axolotl’s immune system upon metamorphosis (Tsai SL, 2020; Ussing and Rosenkilde, 1995), a phenomenon also seen in other amphibians (Pollet, 2010; Robert and Ohta, 2009), could be a key factor in this context. Prior research has highlighted an upregulation in immune system-related gene expression and heightened immune activity in metamorphic axolotls compared with their neotenic counterparts (Pollet, 2010; Robert and Ohta, 2009; Demircan et al., 2020). This increased immune activity could feasibly suppress Flavobacteriaceae colonization in metamorphic blastema tissue, accounting for the relatively lower levels observed. The interaction between opportunistic bacteria, such as members of the Flavobacteriaceae family, and the immune system, particularly within the context of regeneration, presents a compelling area of study. Nevertheless, our existing dataset lacks the capacity to definitively determine the consequences of this communication on the regenerative ability of the axolotl. To gain a deeper understanding of this connection, additional research efforts are crucial, with a specific focus on the dynamic interplay between the host’s immune response and microbial populations throughout the regeneration process. This line of investigation has the potential to unveil crucial insights into the underlying mechanisms of regeneration, offering possible therapeutic targets or approaches to improve regenerative outcomes in various species, including humans.
Our findings also revealed that the Oxalobacteraceae family emerged as a biomarker in neotenic axolotl limb samples at 7DPA, alongside the Flavobacteriaceae family, as indicated by LEfSe analysis. This prominence suggests a potential role in the neotenic axolotl’s limb regeneration process. The Oxalobacteraceae, known for their metabolic activities, including oxalate degradation, may contribute to the regenerative process through the production of beneficial metabolites or modulation of local immune responses (Abratt and Reid, 2010; Asselman et al., 2003; Miller and Dearing, 2013). Their presence in the regenerating limb tissue could also reflect broader interactions within the microbial community that impact tissue repair and regeneration. However, the specific mechanisms underlying the potential involvement of Oxalobacteraceae in axolotl limb regeneration remain speculative. This underlines the need for targeted functional studies to elucidate their exact role and interaction with the host during the regeneration process.
Taken together, our study not only aligns with existing literature on axolotl microbiome dynamics but also introduces new dimensions to our understanding of microbial involvement in limb regeneration. The potential functional roles of specific microbial taxa, such as Flavobacteriaceae, in the regenerative process open avenues for further research, potentially illuminating the intricate microbial–host interactions that underpin regeneration in axolotls and possibly other species.
Conclusions
The current investigation reports a novel examination of the microbiome dynamics linked to axolotl limb regeneration, elucidating the intricate interaction between microbial communities and the regeneration processes during neotenic and metamorphic phases. Our results demonstrate notable alterations in microbial structure, particularly the prevalence of Flavobacteriaceae within the blastema tissue, emphasizing the potential significance of specific microbial taxa in the regeneration mechanism. This research not only yields valuable understandings into the distinctive microbial environment of axolotl regeneration but also emphasizes the necessity for additional functional investigations to elucidate the precise functions of these microbes, such as the families Oxalobacteraceae and Aeromonadaceae, in the regenerative process. The observed variances in microbial profiles between neotenic and metamorphic axolotls, along with their potential implications for regeneration, present a promising direction for future studies in regenerative medicine and microbiology.
Footnotes
Acknowledgments
The author(s) would like to thank their laboratory staff for their assistance in collecting, storing, and managing the samples.
Authors’ Contributions
T.D. conceived and designed the experiments. S.G. and E.A.T. carried out animal experiments and collected the samples. All authors contributed to the amplicon sequencing libraries generation. T.D. analyzed the data, constructed the figures, and wrote the article. All authors have made a significant intellectual contribution and read and agreed to the final published version of the article.
Author Disclosure Statement
The author(s) declare(s) that no conflicting financial interests exist.
Data availability
The sequence data have been made publicly available in the national center for biotechnology information (NCBI) Sequence Read Archive with accession number PRJNA1046296.
Funding Information
This study was financially supported by the Scientific and Technological Research Council of Turkey (TUBITAK) with project number 118S376.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.