
Abstract
The traditional way of thinking about human diseases across clinical and narrow phenomics silos often masks the underlying shared molecular substrates across human diseases. One Health and planetary health fields particularly address such complexities and invite us to think across the conventional disease nosologies. For example, tuberculosis (TB) and lung cancer (LC) are major pulmonary diseases with significant planetary health implications. Despite distinct etiologies, they can coexist in a given community or patient. This is both a challenge and an opportunity for preventive medicine, diagnostics, and therapeutics innovation. This study reports a bioinformatics analysis of publicly available gene expression data, identifying overlapping dysregulated genes, downstream regulators, and pathways in TB and LC. Analysis of NCBI-GEO datasets (GSE83456 and GSE103888) unveiled differential expression of CEACAM6, MUC1, ADM, DYSF, PLOD2, and GAS6 genes in both diseases, with pathway analysis indicating association with lysine degradation pathway. Random forest, a machine-learning-based classification, achieved accuracies of 84% for distinguishing TB from controls and 83% for discriminating LC from controls using these specific genes. Additionally, potential drug targets were identified, with molecular docking confirming the binding affinity of warfarin to GAS6. Taken together, the present study speaks of the pressing need to rethink clinical diagnostic categories of human diseases and that TB and LC might potentially share molecular substrates. Going forward, planetary health and One Health scholarship are poised to cultivate new ways of thinking about diseases not only across medicine and ecology but also across traditional diagnostic conventions.
Introduction
One Health and planetary health are emerging transdisciplinary fields of scholarship that acknowledge the interdependence of human and nonhuman animal health as well as the need to think across the disease diagnostic categories (Mumford et al., 2022). Certain diseases differ in the final clinical presentations but might share underlying molecular substrates. It is well known that tuberculosis (TB) and lung cancer (LC) are two important respiratory disorders with high morbidity and mortality worldwide (Uchiyama et al., 2021). Though these two conditions have diverse pathogenesis, they often coexist in a patient, making the differential diagnosis challenging for the clinicians (Varol et al., 2014; Sun et al., 2023). In addition to that, these overlapping characteristics frequently lead to serious complications in these patients (Bhatt et al., 2012).
Gene expression-based studies play a major role in highlighting how genetic predispositions contribute to disease phenotypes by investigating downstream pathways. Recent bioinformatics research has focused on gene expression patterns in TB (Chen et al., 2023) and LC (Lim et al., 2018). These systems biology and bioinformatics-based studies have discovered altered genes in bronchoalveolar lavage (BAL), blood, and tissue samples obtained from both disorders. It is also important to mention that the predictive capacity can be greatly amplified by integrating machine learning-based algorithms with gene expression data (Dasgupta et al., 2023). Despite the similarities that exist between TB and LC, limited studies have highlighted the common genetic features and associated pathways between these two disorders. Several studies have reported that downstream regulators, including transcription factors (TFs) and microRNAs (miRNAs), may serve as the potential diagnostic biomarkers for TB (Pattnaik et al., 2022) and LC (Li et al., 2013). It is critical to highlight the interaction networks among dysregulated genes, miRNAs, and TFs to comprehend the molecular processes that are associated with both disorders. This information may facilitate the development of more precise diagnostic tools and personalized treatment strategies for patients affected by either or both conditions. Finally, with the rapid emergence of molecular docking, the interactions between key genes and potential therapeutic drugs can be explored (Pinzi and Rastelli, 2019).
An extensive literature search has identified a total of two experimental studies, one retrospective study, and one bioinformatics-based study reported to date involving patients with TB and LC. Zhang et al. analyzed common differentially expressed genes (DEGs) in both diseases, revealing high TLR2 expression. They also concluded that silencing the expression TLR2 may inhibit the migration of tumors in patients with TB-associated LC (Zhang et al., 2019). Another recent experimental study by Barh and his group identified a total of 45 noncoding RNAs (miRNAs) that are significantly dysregulated in both TB and LC (Barh et al., 2018). A retrospective study suggested that the majority of the patients with TB and LC are male having a smoking history (Hu et al., 2020). A recent bioinformatics-based study highlighted the role of two miRNAs, i.e., miR-34a and miR-182, on the pathogenesis of both diseases (Alimardanian et al., 2024). Despite the overlapping clinical associations between TB and LC, no studies have reported the shared genes and associated pathways between these two conditions. It is important to note that analysis of Gene Expression Omnibus (GEO) datasets provides a holistic overview of gene signatures of diverse populations compared with cohort-based studies (Bentley et al., 2017). Studies have also indicated that this bioinformatics-based analysis significantly reduces confounding factors that may arise in cohort-based experiments (Dai et al., 2024; Hayat and Ishrat, 2023).
In the present study, a bioinformatics-based analysis was performed to identify the overlapping dysregulated genes between TB and LC as compared with controls. The expressions of these key genes are also validated in two fresh GEO datasets. Through the integration and analysis of gene expression data, the shared pathways that may offer insights into the intersection of these two pulmonary diseases are highlighted. The miRNAs and TFs that are associated with these overlapping genes are also identified. A machine learning-based random forest model was developed to explore the efficacy of these genes in differentiating disease groups from controls. Finally, the potential drugs that target these genes are indicated, and molecular docking analysis confirmed the binding ability of the identified drugs with the key genes.
Materials and Methods
Datasets section from NCBI-GEO database
The data used in this study were procured from NCBI-GEO tool available at http://www.ncbi.nlm.nih.gov/geo. Two keywords, “tuberculosis” and “lung cancer” were used to screen the data (last accessed: 19.4.2024). To identify the common DEGs between TB and LC, two microarray-based datasets, i.e., GSE83456 and GSE103888, were selected. GSE83456 comprises a total of 45 patients with TB and 61 controls. Whole blood ribonucleic acid (RNA) was collected from the recruited subjects, and 750 ng of complimentary RNA was hybridized to Illumina Human HT-12 V4 BeadChip arrays and scanned on Illumina iScan (Blankley et al., 2016).
The GSE103888 dataset contains BAL cell samples obtained from 13 tumor-located regions and 6 nontumor locations. During standard diagnostic bronchoscopy, BAL specimens were collected from the lung segment where the tumor was located. In contrast, for control cases, BAL was collected from the right middle lobe. The bronchoscopy procedure involved instilling saline water (250 mL) to harvest the cells. Next, RNA was extracted from BAL cells, and microarray analysis was performed using Affymetrix Human Genome U133 Plus 2.0 Array. The baseline characteristics of the recruited subjects are shown in Supplementary Table S1 (Kuo et al., 2018). The present study was conducted under the overall research ethics oversight of the author’s institution. The study design workflow is depicted in Figure 1.
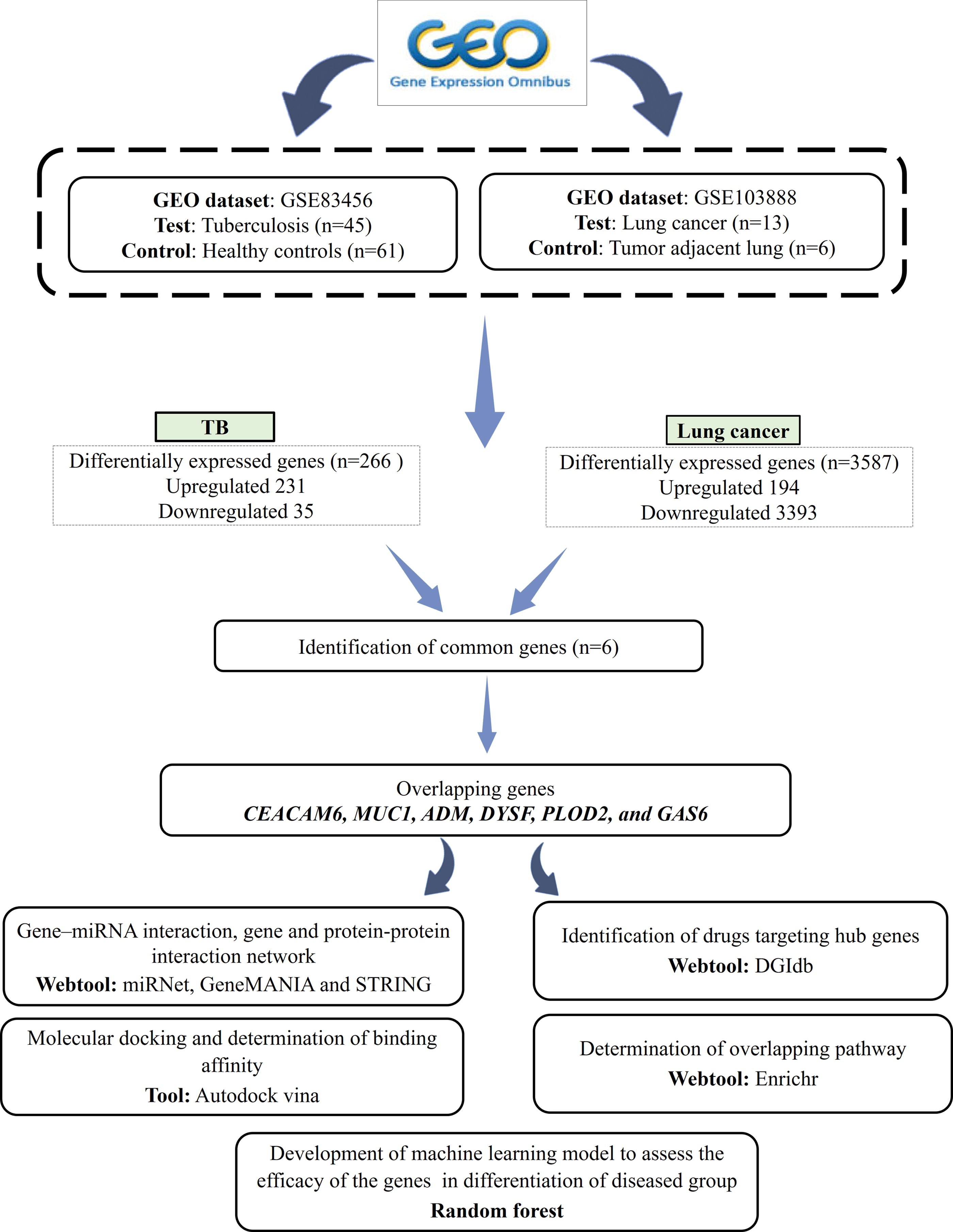
Workflow of the study design. Two Gene Expression Omnibus (GEO) databases were selected to identify the common differentially expressed genes (DEGs) between tuberculosis (TB) and lung cancer (LC).
Screening of DEGs
Data analysis was conducted using the GEO2R web tool. In the panel of the samples, the disease groups were considered as ‘test’ and the healthy subjects were selected as “control” (https://www.ncbi.nlm.nih.gov/geo/info/geo2r.html#groups). GEO2R uses GEOquery and limma to conduct a comparative gene expression analysis. GEOquery facilitates the transformation of GEO-based data into R-compatible structures, and limma provides a robust statistical framework to identify the variations in gene expression within microarray datasets (Ritchie et al., 2015). DEGs for both TB and LC were selected by a threshold|log2 (fold change)|<1 , false discovery rate < 0.10, and p-value <0.05 (Dasgupta et al., 2023).
Similarity analysis of DEGs
DEGs that were commonly altered in both TB and LC as compared with controls were identified. A Venn diagram was plotted to visualize the overlapping DEGs between these two diseases. Additionally, the percentage of shared DEGs compared with the total number of altered genes was calculated. These common DEGs were regarded as prime data points throughout the study.
Enrichment and network analysis
The pathways associated with the common DEGs were determined using EnrichR webtool (http://amp.pharm.mssm.edu/Enrichr). This tool contains a total of 1,80,184 annotated gene sets from various libraries and an advanced search engine that correlates the gene expression data with Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Kuleshov et al., 2016). The enrichment p-value of the associated pathway is also calculated.
Gene and protein–protein interaction network
GeneMANIA (http://www.genemania.org), a popular user-friendly web interface, is used to develop the correlation network and determine the coexpression patterns (Warde-Farley et al., 2010). Next, the search tool for the retrieval of interacting genes/proteins (STRING) tool (https://stringdb.org/cgi/input) was used to create the protein–protein interaction (PPI) network, which illustrates the relationship between the proteins and elucidates the underlying molecular mechanism (Szklarczyk et al., 2019).
DEG–miRNA and TF interaction network
miRNet (https://www.mirnet.ca/), an integrated platform connecting miRNAs and TFs with genes, was used to predict the interaction networks between DEG and miRNAs as well as DEG and TF. Interaction data from miRNet were collected from three databases: TarBase v7.0, miRTarBase v7.0, and miRecords (Chang et al., 2020). Cytoscape (https://cytoscape.org/) was used to visualize DEG–miRNA and DEG–TF regulatory networks (Shannon et al., 2003).
Identification of promising drugs
In the present study, the Drug-Gene Interaction database (DGIdb) was utilized to identify promising drugs targeting the overlapping genes between TB and LC. The database provides information on drugs that target the gene of interest based on various literature sources (https://dgidb.org) (Wagner et al., 2016). Through inputting genes of interest into DGIdb, four potential drugs along with their interaction scores with the key genes were identified, retaining a cut-off interaction score of 0.75.
Molecular docking between drug and key gene
In the present study, AutoDock Vina tool was used to investigate the interactions between key genes and identified drugs by assessing their binding affinity. The three-dimensional structures of the target gene and drug of interest were obtained, and molecular docking analysis was performed. The interaction energy and conformation for possible drug–protein pairs binding were calculated (Trott and Olson, 2010). Further, the docking results were analyzed to detect the most favorable interactions based on the binding affinity and structural compatibility.
Predictive model using random forest classifier
A random forest classifier, with a total of 50 trees, was used for the dimensionality reduction of gene datasets. A predictive model utilizing this classifier was developed to map the scores of feature importance for the common genes and explore their potential in the classification of TB and LC. A multivariate receiver operating characteristic (ROC) curve analysis was also conducted with the classifier. The area under the multivariate ROC curve was calculated using the scikit-learn package in Python 3.8 (https://scikit-learn.org/stable) (Ghosh et al., 2021).
Differential diagnosis of TB and LC
A supervised classification model, orthogonal partial least squares discriminant analysis (OPLS-DA) was developed to highlight the differences between TB and LC using MetaboAnalyst (v5.0) (https://www.metaboanalyst.ca/) (Pang et al., 2021). R2 which corresponds to the goodness of fit and Q2 which indicates the predictive ability of the model were calculated to explore the robustness of the generated models. Variable importance in projection (VIP) score plot, which ranks the gene features based on their significance in differentiating TB and LC, was developed (Triba et al., 2015).
Results
Screening of DEGs in TB and LC
After processing raw data, a total of 266 DEGs, including 231 upregulated and 35 downregulated genes, were found in TB patients as compared with controls. In the case of LC, 194 elevated and 3393 downregulated genes were identified when compared with controls. Supplementary Tables S2 and S3 display the 10 most significantly altered genes in both diseases.
Six common DEGs identified between TB and LC
A total of 266 DEGs for TB and 3587 DEGs for LC were compared, and six common genes (CEACAM6, MUC1, ADM, DYSF, PLOD2, and GAS6) were found to be differentially expressed in both diseases as compared with controls. These genes were found to be significantly upregulated in both TB and LC patients. Venn diagram in Supplementary Fig. S1 demonstrates the common DEGs between the two diseases, indicating that the common DEGs comprise 0.15% of the total 3853 DEGs.
Validation of the overlapping genes
An extensive literature search was carried out, and GSE262613 and GSE168198 were selected to validate the expression of the six common DEGs in TB and LC, respectively. In both datasets, CEACAM6, MUC1, PLOD2, ADM, and GAS6 were found to be upregulated in TB and LC as compared with controls. It is also important to mention that inconsistencies may arise while interpreting the result as various cofactors, including study design, experimental procedure, and subject characteristics, may differ between two datasets. However, the electronic and literature-based validation of the six genes strengthens the findings and suggests that these genes are commonly associated with the pathogenesis of both TB and LC.
Interaction network and enriched pathway
GeneMania interaction network exhibited 88.47% coexpression and 11.53% colocalization between the common genes (Fig. 2A). The PPI network that was constructed using the STRING online tool indicates a total of 11 nodes and 9 edges present between the connected proteins (Fig. 2B). Next, the gene set enrichment analysis was conducted using Enrichr tool. It was found that lysine degradation was significantly enriched in both TB and LC (p-value = 0.01) (Fig. 2C).
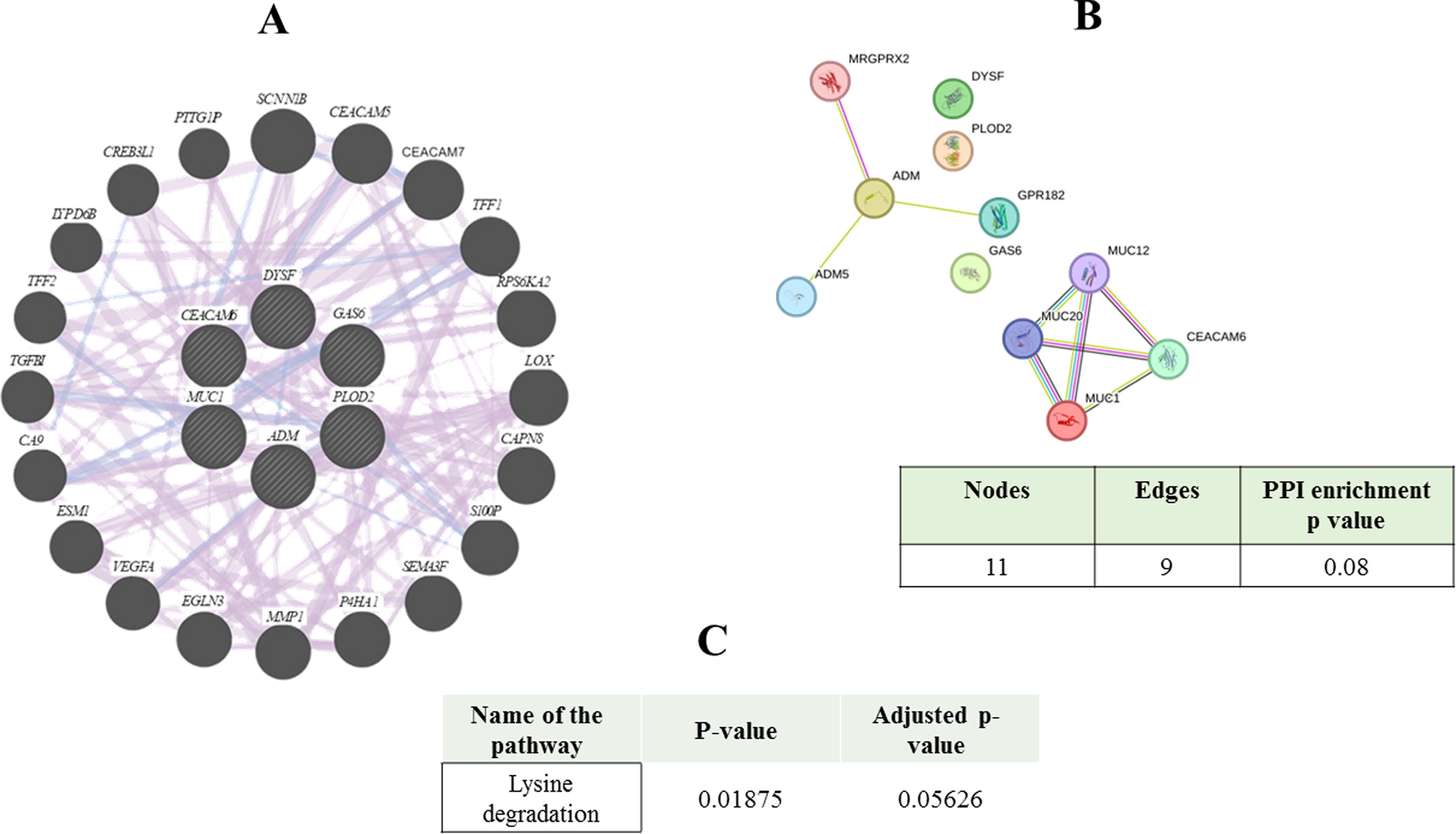
Interaction network and enriched pathway associated with the common genes between tuberculosis (TB) and lung cancer (LC).
Identification of potential miRNAs and TFs
A total of three miRNAs, i.e., hsa-miR-203a-3p, hsa-miR-124-3p, and hsa-miR-155-5p, showed the highest association with the common genes based on the degree centrality of the gene–miRNA network (cut-off value = 4). The degree centrality highlights the number of connections present in a single node. The complete network comprises a total of 246 nodes and 312 edges (Supplementary Fig. S2). Furthermore, the miRNet web-based tool revealed that three TFs, i.e., AR, GATA1, and GATA3, exhibited the highest interaction with the six common DEGs between TB and LC. The cut-off value for degree centrality was selected as 1 for this analysis.
Identification of potential drugs and molecular docking
The DGIdb webtool indicated that warfarin has the highest interaction with one of the key genes, GAS6. The drug has an interaction score of 1.72 and a query score of 0.24 with this common gene between TB and LC. The list of additional promising drugs that can target the other common genes is shown in Supplementary Table S4. Next, to explore the interaction between GAS6 and warfarin, molecular docking analysis was performed. The RSCB-PDB (https://www.rcsb.org/) database was used to obtain the PDB structure of GAS6 (PDB ID: 1h30). The three-dimensional structure of the candidate drug, i.e., warfarin, was retrieved from the PubChem tool (https://pubchem.ncbi.nlm.nih.gov/). Using AutoDock Vina, the receptor and the ligand were prepared for molecular docking analysis by adding hydrogens and Gasteiger charge. The docking analysis revealed a strong binding affinity between GAS6 and warfarin, i.e., −7.1 kcal/mol (Fig. 3).

Molecular docking shows the binding affinity between GAS6 and warfarin drug. The binding affinity score between the key protein and drug is −7.1 kcal/mol.
Evaluation of common genes using random forest classifier
The results of the multivariate ROC analysis indicate that the overall area under the curve (AUC) for the common genes in distinguishing between TB and controls is 0.901 and for LC and controls is 0.900 (Fig. 4A and B). The model exhibited accuracies of 84% and 83% in discriminating TB and LC from controls, respectively. Additionally, the relative importance of the six common genes is illustrated in Figure 4C and D; MUC1 and DYSF showed the highest feature importance in distinguishing TB and LC from controls.
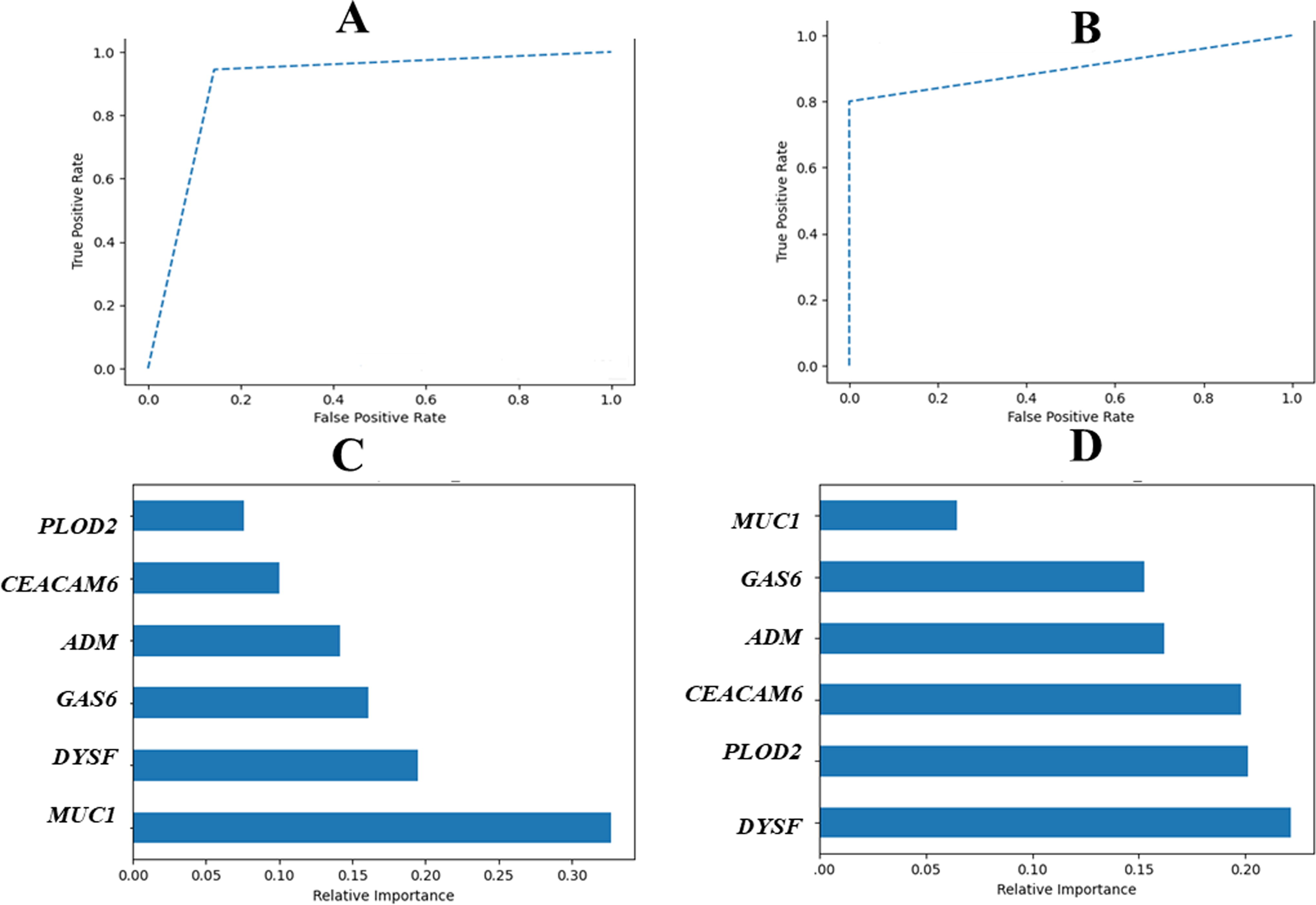
Development of classification models for tuberculosis (TB) and lung cancer (LC) from controls.
Multivariate model for classification of TB and LC
The OPLS-DA model exhibits a clear demarcation between the two disease groups based on the gene expression pattern. The robustness of the model is evaluated based on the R2 and Q2 values (R2X: 0.921, R2Y: 0.98, and Q2: 0.98). Finally, the VIP plot exhibited that FTL, RPL19, CTSC, CD81, and TMEM59 are among the top five genes contributing significantly to the differentiation of TB and LC (Fig. 5). The VIP scores of these top five discriminatory genes are provided in Supplementary Table S5.

The supervised classification orthogonal partial least squares discriminant analysis (OPLS-DA) model showing differences between tuberculosis (TB) and lung cancer (LC).
Discussion
The identification of shared genes between TB and LC is a pressing need considering the rise of One Health and planetary health that invites the life sciences and medical research community to think across the traditional clinical disease silos (Lusiki et al., 2023). The present study is the first of its kind that reveals the common as well as differential genes between these two chronic pulmonary disorders. This study uncovers that CEACAM6, MUC1, ADM, DYSF, PLOD2, and GAS6 are altered in both TB and LC patients as compared with controls. Furthermore, the lysine degradation pathway shows significant association with both disorders. miRNAs, including hsa-miR-203a-3p, hsa-miR-124-3p, and hsa-miR-155-5p, and three TFs, such as AR, GATA1, and GATA3, exhibited an association with the overlapping genes between TB and LC. Warfarin exhibited the highest potential in targeting one of the key overlapping genes, GAS6. In addition to the overlapping genes, the present study also sheds light on the differences between these two diseases; FTL, RPL19, CTSC, CD81, and TMEM59 exhibited the highest potential in the classification of TB from LC. Overall, this study contributes significantly to redefining our approach to disease classification and treatment strategies by emphasizing the interconnectedness of diseases and the importance of interdisciplinary research within the One Health and planetary health paradigms.
The present study indicates an upregulated expression of CEACAM6, MUC1, ADM, DYSF, PLOD2, and GAS6 in both TB and LC. The increased expression of CEACAM6 may be attributed to the body’s defense mechanism against “Mycobacterium tuberculosis” pathogens in TB. In the case of LC, the upregulation of this gene contributes to cancer progression by aiding tumor cell adhesion and migration (Rizeq et al., 2018). MUC1, a gene that encodes membrane-bound proteins, is also found to be increased in both TB and LC. Similar to the previous gene, the alteration of MUC1 is also associated with host defense mechanisms against the pathogen in patients with TB (Inoue et al., 1995). The upregulated expression of MUC1 contributes to tumor proliferation and metastasis in patients with LC (Chen et al., 2021). In TB, ADM alteration assists in managing the inflammatory response and vascular dynamics during infection. The angiogenic property of ADM is also documented in LC (Chen et al., 2011). DYSF plays a key role in muscle repair and inflammatory pathways, which are the hallmarks of both TB and LC (Bansal and Campbell, 2004). Next, the increased expression of PLOD2 is reported to be associated with tissue remodeling and extracellular matrix modifications in cancer (Cheriyamundath et al., 2021). GAS6, which is mainly involved in cell proliferation, is found to be overexpressed in both TB and LC. In TB, GAS6 influences the immune regulation and inflammation. In the case of LC, the upregulated expression supports tumor growth and proliferation (Laurance et al., 2012).
The enrichment pathway analysis indicated that the lysine degradation pathway is significantly associated with the pathogenesis of both TB and LC. Studies have reported that lysine plays a major role in tumor formation during the development of LC (Hsu et al., 2021). In the case of TB, lysine degradation has been correlated to the production of metabolites that influence the ability to regulate the pathogens (Gokulan et al., 2003). Besides the overlapping genetic features, the multivariate classification model indicated the five genes responsible for the differentiation of TB and LC. It was found that FTL, RPL19, CTSC, CD81, and TMEM59 have the maximum potential in the classification of TB from LC. It is reported that FTL and CTSC genes are mainly involved in the metastatic pathway of cancer (Chen et al., 2017; Kim et al., 2024). On the other hand, RPL19 is mainly involved in the interferon-gamma signaling pathway and progression of tumors in cancer (Kuroda et al., 2010). The role of CD81 and TMEM59 is documented in TB patients as they are actively involved in granuloma formation, which is an important hallmark of TB (Zheng et al., 2022; Ullrich et al., 2010).
miRNAs, such as hsa-miR-203a-3p, hsa-miR-124-3p, and hsa-miR-155-5p, are found to be significantly associated with the overlapping genes between TB and LC. In recent years, several miRNAs have shown their effectiveness in the regulation of gene expression and progression of disease (Ying et al., 2020). It is also envisioned that miRNAs could be promising biomarkers for several pulmonary disorders. Shi et al. highlighted the role of hsa-miR-203 in chronic obtructive pulmonary disease (COPD), another inflammatory pulmonary disorder. The authors reported that miR-203 miRNA acts as an immune response inhibitor and targets TAK1 and PIK3CA genes in these patients. Their findings indicated that miR-203 could be a prognostic biomarker of COPD (Shi et al., 2015). Interestingly, a recent study by Sanchez-Cabrero et al. indicated that miR-124, present in both exosomes and free-circulation, is significantly upregulated throughout the progression of nonsmall cell LC. According to this study, miR-124 could be a potential biomarker for both advanced and early-staged patients (Sanchez-Cabrero et al., 2023). Another miRNA, miR-155, also exhibited a strong association with the common genes between TB and LC in the present study. Consistent with this observation, Ying et al. suggested that miR-155 could be an effective biomarker of active pulmonary TB as it is significantly upregulated in the sputum of these patients (Ying et al., 2020). A total of three TFs (AR, GATA1, and GATA3) displayed an association with the common genes. AR is reported to be involved in metastatic pathways in LC and granuloma formation in TB (Liu et al., 2023). The two other TFs, i.e., GATA1 and GATA3, are reported to be the key players of inflammatory pathways and cell proliferation in both TB (Oguz et al., 2008) and LC (Wang et al., 2023).
Warfarin, a well-known anticoagulant, has been found to have the potential to target GAS6, a gene that plays a key role in both TB and LC. As it is reported that GAS6 is involved in promoting tumor growth and metastasis in LC, suppression of this gene could be a promising therapeutic approach for treating these patients (Gomes et al., 2019). Moreover, GAS6 plays a major role in modulating the host immune response in patients with TB (Wang et al., 2021). These reports indicate that targeting GAS6 with the promising drug warfarin could be effective in treating LC patients with a history of TB.
This study has several limitations. First, the two datasets used in this study are distinct; one involves blood samples from TB patients, whereas the other comprises BAL cells from LC groups. Second, the biological processes related to the common genes are limited by bioinformatics methods and remain to be proven by experimental studies. Third, several factors, including age, sex, race, environmental factors, and smoking status, were not adjusted into the analysis of DEGs. Multicentric experimental studies across the country/different continents/countries are recommended to validate the potential of these overlapping as well as differential genes.
Conclusions
In summary, the present study offers a novel insight into the pathogenesis of both TB and LC. A total of six genes (CEACAM6, MUC1, ADM, DYSF, PLOD2, and GAS6) are found to be altered in both TB and LC as compared with controls. Furthermore, the lysine degradation pathway is found to be enriched in both disorders. The miRNAs (hsa-miR-203a-3p, hsa-miR-124-3p, and hsa-miR-155-5p) and TFs (AR, GATA1, and GATA3) associated with the key genes are also documented. Furthermore, the present study indicated that warfarin could be a potential drug that targets one of the key genes, GAS6. Whereas warfarin is not conventionally regarded as a novel drug for treating TB and LC, this study proposes its potential in treating LC patients with a history of TB or TB-associated LC patients. Furthermore, the genes that can differentiate TB from LC (FTL, RPL19, CTSC, CD81, and TMEM59) are also identified. In all, this study provides a new direction in the field of exploring the shared and differential genetic signatures between two overlapping pulmonary disorders, TB and LC.
Footnotes
Acknowledgments
The author is thankful for the permission to use the freely available figure-making tool BioRender.com.
Author’s Contribution
S.D.: Conceptualization, formal analysis, investigation, visualization, and writing—review/editing.
Author Disclosure Statement
The author declares that she has no conflicting financial interests.
Funding Information
No funding was received for the present study.