
Abstract
Rare diseases and conditions have thus far received relatively less attention in the field of precision/personalized medicine than common chronic diseases. There is a dire need for orphan drug discovery and therapeutics in ways that are informed by the precision/personalized medicine scholarship. Moreover, people with rare conditions, when considered collectively across diseases worldwide, impact many communities. In this overarching context, Activin A Receptor Type 1 (ACVR1) is a transmembrane kinase from the transforming growth factor-β superfamily and plays a critical role in modulating the bone morphogenetic protein signaling. Missense variants of the ACVR1 gene result in modifications in structure and function and, by extension, abnormalities and have been predominantly linked with two rare conditions: fibrodysplasia ossificans progressiva and diffuse intrinsic pontine glioma. We report here an extensive bioinformatic analyses assessing the pool of 50,951 variants and forecast seven highly destabilizing mutations (R206H, G356D, R258S, G328W, G328E, R375P, and R202I) that can significantly alter the structure and function of the native protein. Protein–protein interaction and ConSurf analyses revealed the crucial interactions and localization of highly deleterious mutations in highly conserved domains that may impact the binding and functioning of the protein. cBioPortal, CanSAR Black, and existing literature affirmed the association of these destabilizing mutations with posterior fossa ependymoma, uterine corpus carcinoma, and pediatric brain cancer. The current findings suggest these deleterious nonsynonymous single nucleotide polymorphisms as potential candidates for future functional annotations and validations associated with rare conditions, further aiding the development of precision medicine in rare diseases.
Introduction
A type I receptor for the ligands of the transforming growth factor (TGF)-family, activin A receptor type I (ACVR1), is also known as activin receptor-like kinase 2 (ALK2). ACVR1 (UniProtKB: Q04771) is a 509 amino acid protein encoded by the human ACVR1 gene (Ensembl: ENSG00000115170) (Boutet et al., 2007; Zerbino et al., 2018) found on chromosome 2q24.1. ACVR1 is a single-pass type I membrane protein predominantly localized in the plasma membrane within the cell, with its subcellular localization specified to nucleoli and cytosol (Shore et al., 2006; Thul and Lindskog, 2018). Recent research has revealed that several disorders are linked to the genetic alteration of the ACVR1 gene.
The first and earliest known disorder linked to the ACVR1 gene was fibrodysplasia ossificans progressiva (FOP) (OMIM #135100), an autosomal dominant rare congenital disorder with a recurrent mutation (ACVR1R206H) associated with progressive heterotopic ossification of ligaments, tendons, and skeletal muscle. In the coding exon 4 of the ACVR1 gene, at c.617G > A, the substitution of amino acid at position 206 from arginine to histidine (p.206H), a missense mutation with identical heterozygosity, is reported in more than 90% of FOP patients. The ACVR1 p.R206H mutation makes this receptor more responsive to ligands. It is located at the end of the glycine-serine (GS) domain, resulting in a gain of function mutation in the type I TGF-β receptor (Katagiri et al., 2021). However, mutations in ACVR1 gene’s coding region are also reported and linked to diffuse intrinsic pontine glioma (DIPG) (Buczkowicz et al., 2014; Fontebasso et al., 2014; Taylor et al., 2014), diffuse idiopathic skeletal hyperostosis (DISH) (Gupta et al., 2019), posterior fossa ependymoma (PFE) (Pratt et al., 2022), and congenital heart defects (CHD) (Smith et al., 2009).
The ACVR1 protein has a single peptide, an N-terminal ligand-binding domain (extracellular), followed by a transmembrane domain, a GS-rich domain (intracellular), and a protein kinase (PK) domain, respectively. The crucial residues necessary for phosphorylating proteins to activate ACVR1 are found in the loop of the GS domain (Nagar et al., 2023). ACVR1 is associated with type II receptors, such as ACVR2A, ACVR2B, and BMPR2, which form a heterotetrameric receptor complex. Initially, it was known that ACVR1 binds with activin A and, on complex formation with BMP type II receptors, triggers SMAD2/3 activation and phosphorylation.
It was identified later that receptor ACVR1 binds particularly to osteogenic ligands like BMPs or activin A with different affinities that, in turn, trigger intracellular signaling through Smad1/5/8 and Smad 2/3 phosphorylation. Interestingly, ACVR1 can also be activated by noncanonical signaling pathways in addition to the canonical Smad signaling (Valer et al., 2019).
Moreover, KEGG pathway database and Human Protein Atlas were accessed to view the molecular pathways (osteogenic differentiation, iron metabolism, neurogenesis, and placenta formation in embryonic differentiation) and expression summary of ACVR1 that is highest in endocrine-thyroid, parathyroid, adrenal, and pituitary glands and muscle tissues. The variable concentration of ACVR1 expression was observed among different body tissues. Among them, the thyroid gland and smooth muscle showed the highest expression level contrary to its lowest in the ovary, adipose tissue, appendix, spleen, lymph node, tonsil, and soft tissue, as depicted in Supplementary Figures S2 and Figure S3. Highest RNA expression of ACVR1 was reported in thyroid gland and smooth muscles, followed by endometrium and cervix, while the lowest was in the retina, testis, and bone marrow, as illustrated in Supplementary Figure S4 (Kanehisa et al., 2017; Thul and Lindskog, 2018). ALK2/ACVR1 has been the subject of extensive research over the past 15 years due to recognizing the direct link of its gain-of-function mutations contributing to the development of various disorders, viz., FOP, DISH, DIPG, PFE, and CHD (Katagiri et al., 2021).
Single nucleotide polymorphisms (SNPs) are the most prevalent genetic variations associated with significant disease risk and serve as a clear candidate marker in genome-wide association studies. SNPs contribute to 90% of the known human genetic variations to date. They are caused by a germline point mutation at a specific location possessing an alternative base within the locus, giving rise to different alleles. This change may further impact the protein’s structure, function, folding, stability, interaction, charge distribution, and dynamics with other proteins. The nonsynonymous SNPs (nsSNPs) may cause a significant amino acid substitution and result in protein functional diversity, causing a single base substitution in its coding region. Such missense or deleterious variants, in turn, modify the encoded amino acids and impact the structural and functional integrity of the protein. Understanding of the disease-causing variants and their association with the phenotypes can be foreseen for future advancement in personalized treatments (Cargill et al., 1999; Collins et al., 1998; Lander, 1996).
It is complex and extortionate to investigate the impact of nsSNPs on protein’s structure and function. Hence, predicting deleterious nsSNPs by computational tools and algorithms is an inexpensive and decisive alternative (Yazar and Özbek, 2021).
In the field of precision medicine, rare diseases and conditions have received relatively less attention than common chronic diseases. This research and knowledge gap in precision medicine ought to be addressed, first, on principled grounds for health for all, and second, and not least, because people with rare conditions, when considered collectively across the diseases worldwide, represent and impact many communities. Hence, the present study (Supplementary Fig. S1) aimed to explore the in silico algorithms and tools so as to forecast the structural and functional implications of nsSNPs on ACVR1 and the phenotypic impact description, structural or functional features, and pathogenicity of the variant associated with diseases.
Materials and Methods
The present investigation in silico did not involve the recruitment of human subjects or animal subjects and did not require informed consent and institutional review board approval. The study was conducted under the overall research ethics oversight of the authors’ institutions.
ACVR1 SNP data collection and variant effect prediction
The nsSNPs linked to the human ACVR1 gene were retrieved from the dbSNP database (Sherry et al., 2001). A total of 50,951 variants were retrieved and processed with the variant effect prediction (VEP) tool to determine their impact on the ACVR1—gene, transcript, protein, and regulatory regions. Furthermore, the protein structure and sequence of ACVR1 were obtained from the Protein Data Bank (PDB ID—6I1S) (Fortin et al., 2020; Sussman et al., 1998) and UniProt (UniProtKB-Q04771) (Boutet et al., 2007), respectively. MODELLER and PyMOL tools were utilized to fill the missing residues and create the mutation models (Briggs et al., 2002; Eswar et al., 2008).
Sequence-based prediction of functional consequences of nsSNPs
The impact of ACVR1-linked missense mutation was predicted by utilizing various sequence-based computational tools—SIFT, PolyPhen-2, PROVEAN, PANTHER-PSEP, SNAP2, and PhD-SNPg. SIFT (sorting intolerant from tolerant) predicts the amino acid substitution impact as intolerant (deleterious) or tolerant (neutral) based on the physical properties (score >0.05; neutral and score <0.05; deleterious) (Kumar et al., 2009; Sim et al., 2012). Polyphen-2 (polymorphism phenotype-2) is a naive Bayes classifier (cutoff value 1.0) that utilizes comparative and physical parameters to calculate the position-specific independent substitution score (score: 0.4–0.9; possibly damaging, score >0.9; probably damaging, and score <4.0; benign) (Adzhubei et al., 2013).
PROVEAN (Protein Variation Effect Analyzer) uses the delta score prediction (cutoff value of −2.5) based on protein sequence homology and variant sequence alignment (score ≤−2.5; deleterious and score >−2.5; neutral) (Choi and Chan, 2015). PANTHER-PSEP (position-specific evolutionary preservation) analyzes the functional consequences of variation using ancestral sequence reconstruction and evolutionary conservation time calculations (score: 0–1; probability of deleterious and score >0.5, disease-causing) (Tang and Thomas, 2016). SNAP2 (screening for nonacceptable polymorphisms) is a neural network-based algorithm that predicts the functional effect of amino acid substitution on proteins (score range: –100 to +100, highly neutral to highly effective) (Hecht et al., 2015). PhD-SNPg is a gradient-boosting algorithm-based binary classifier (score: 0–1) that predicts the pathogenicity index of a variant (score >0.5, significantly pathogenic) (Capriotti and Fariselli, 2017). The predicted variants list was used for Jvenn viewer to identify the overlapping deleterious ones (Bardou et al., 2014).
Structure-based prediction of functional consequences of deleterious
The impact of nonsynonymous SNPs of ACVR1 was analyzed for their functional and structural consequences using computational tools like DUET, DynaMut, mCSM, SDM, and CUPSAT. DUET uses support vector machine for a consolidated computational approach (SDM and mCSM) to predict the effect of mutation on the stability of the protein (Pires et al., 2014a). DynaMut uses a normal mode and graph-based signature approach to predict protein stability by assessing the vibrational entropy change (Rodrigues et al., 2018). The mCSM web server uses a framework of graph-based signature and energetic and evolutionary information to predict the mutation’s impact on a protein’s structure (Pires et al., 2014b). The SDM (Site Directed Mutator) web server is a function of statistical potential energy used to assess the effect of mutation on protein stability. It evaluates the stability score using environment-specific variation frequency in homologous protein families (Pandurangan et al., 2017). CUPSAT (Cologne University Protein Stability Analysis Tool) uses torsion angle potential and chemical properties based on atom potentials to predict and analyze the changes in protein stability due to single amino acid substitution (Parthiban et al., 2006).
High-risk nsSNPs conservation profile analysis
Conservation analysis offers a deeper understanding of residual evolution, which critically impacts the structure and function of proteins. The ConSurf server predicts evolutionary and phylogenetic conservation by identifying amino acid residues’ structural and functional impact based on the Empirical Bayesian scoring ranging from variable to conserved region (1 to 9), that is, from low to high. Structural analysis of ACVR1 protein delineates its highly conserved residues, which were further indicated as functional and structural if exposed and buried, respectively. If a mutation occurs at a highly conserved residue, it will likely impact a protein’s structure and function and can be determined as damaging in nature. (Ashkenazy et al., 2016).
Interaction network analysis
Owing to their impact on the protein–protein interfaces, several nsSNPs may be disease causing. Findings suggest that, compared to random distribution, the nsSNPs causing disease are often found at the interface of protein–protein interaction (PPI) rather than at noninterface surface regions (David et al., 2012). GeneMANIA (Multiple Association Network Integration Algorithm) (Mostafavi et al., 2008) and STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) (Mering, 2003) databases were used to predict the PPI network for ACVR1. It utilizes a variety of data and resources, including colocalization information, physical interaction information, coexpression information, functional relationships, pathway information, genetic interaction, and protein domain data (Warde-Farley et al., 2010).
Prediction of SNPs’ impacts on ACVR1
The effect of deleterious SNPs on the 3D structure of the ACVR1 protein was explored using Project HOPE [Have (y)Our Protein Explained]. HOPE, an open web server, utilizes several information sources to create homology models by the YASARA program for carrying out necessary functions. HOPE utilizes the FASTA format of a protein to predict the structural impact of point mutation on the protein compared with its wild type (WT) (Venselaar et al., 2010).
Molecular dynamic simulation analysis
To investigate the dynamic stability of both mutated and WT ACVR1 proteins, molecular dynamics (MD) simulation was conducted using GROMACS version 2020. The simulation setup involved an orthorhombic box with a buffer of 10 × 10 × 10 Å, using the TIP3P (transferable intermolecular potential with 3 points) solvent model for system solvation. The CHARMM36 force field was utilized to parameterize the system. To ensure system neutrality, Na+ and Cl− ions were added. The system was then subjected to energy minimization to remove any steric clashes or bad contacts. It was followed by equilibration in two phases as follows: a 10 ns NVT (constant number of particles, volume, and temperature) simulation to stabilize the temperature and a 10 ns NPT (constant number of particles, pressure, and temperature) simulation to stabilize the pressure. During the NVT equilibration, the system temperature was maintained at 310 K using the V-rescale thermostat with a coupling constant of 0.1 ps. For the NPT equilibration, the pressure was maintained at 1 bar using the Parrinello-Rahman barostat with a coupling constant of 2.0 ps.
Postequilibration, the production of MD run was performed for 400 ns. The time step for integrating equations of motion was set to 2 fs. The Particle-Mesh Ewald method was used for long-range electrostatic interactions, with a cutoff of 1.2 nm for both Coulombic and van der Waals interactions. Indexing was done around the 5 Å of the mutated protein. The trajectories obtained from the simulations were analyzed using QTGrace, providing detailed insights into the structural dynamics and stability of WT and mutated ACVR1 proteins (Abraham et al., 2015; Hess et al., 1997).
Essential dynamics analysis
Principal component analysis (PCA) is widely utilized to calculate essential dynamics in MD simulations as it effectively reduces the complexity and dimensionality of the data. The carbon-alpha (Cα) atoms from 4001 frames generated by the 400 ns MD simulation trajectory were aligned to a reference structure to remove overall translational and rotational motions to perform PCA calculations. This alignment is crucial for ensuring that the analysis focuses on the internal motions of the protein. PCA was then computed using the built-in ′covar′ and ′anaeig′ scripts in the GROMACS package. The ′covar′ script calculates the covariance matrix, which consists of atomic fluctuations, by analyzing the positional deviations of the Cα atoms.
The essential files required for performing PCA include the trajectory file (′traj.xtc′), the topology file (′topology.tpr′), and the index file (′index.ndx′). The covariance matrix, which captures the correlated motions of atoms, is diagonalized to obtain the eigenvalues and eigenvectors. Eigenvalues represent the magnitude of variance along each principal component, whereas eigenvectors describe the direction of these motions. The principal components capturing the most significant conformational changes in the protein structure during the simulation can be determined by examining the eigenvalues and eigenvectors. Typically, the first few principal components (those with the largest eigenvalues) account for the majority of the motion, allowing us to focus on these components for detailed analysis (Abraham et al., 2015).
Free energy landscape analysis
Following the molecular dynamics simulation, free energy landscape (FEL) analyses were conducted to gain insights into the collective motions of ACVR1 and its mutations. FEL analysis is instrumental in understanding how a protein transitions between different energy states, providing valuable insights into its stability, folding, and function. The following formula can be used to construct the FEL:
Where ΔG (X) represents the free energy, KB is the Boltzmann constant, T is the absolute temperature, and P (X) is the probability distribution of the principal components of the molecular system. PCA was used to derive the primary components (PC1 and PC2) from the trajectories of the Cα atoms. These components capture the most significant motions within the protein. The FEL analysis was performed using a custom Python script that processed the GROMACS trajectory files. The script utilized the PC1 and PC2 data from the PCA to calculate the FEL, ensuring a detailed and accurate representation of the protein’s energy landscape. By examining the FELs, we could discern the stability and conformational behavior differences between the WT ACVR1 and its mutations.
Identifying disease association of deleterious ACVR1 nsSNPs
A multidimensional cancer genomics database, cBioPortal (Gao et al., 2013), was accessed to explore the cancer genomics data related to the ACVR1 gene; the lollipop plot was utilized to determine the distribution and association of its nsSNPs with cancer. CanSAR Black is an integrative drug discovery and translational research knowledge base (Coker et al., 2019). The mutation profile of this database was also explored to identify the association of deleterious ACVR1 nsSNPs with specific cancers.
Results and Discussion
ACVR1 SNP data collection and variant effect prediction
In total, 50,951 SNPs were obtained for the human ACVR1 gene from the dbSNP database, among which 49,168 were intronic, 1858 were noncoding transcript variants, 339 were missense (nsSNPs), 205 were synonymous variants, 5 were inframe indel, 2 were inframe insertion, 1 was initiator codon variant, and one was inframe deletion. The VEP analysis showed that ACVR1 SNPs overlapped with 4 genes, 29 transcripts, and 44 regulatory features. Based on the prediction consequences (Fig. 1a), 83% of SNPs were in the noncoding intronic region, and 58% of SNPs in the coding region were missense variants.

Sequence-based prediction of functional consequences of nsSNPs
The 339 nsSNPs of ACVR1 were used in SIFT, Polyphen-2, PROVEAN, PANTHER-PSEP, SNAP2, and PHD-SNPg for predicting the sequence-based impact of nsSNP on its function. SIFT predicted 9, Polyphen-2 predicted 14, PROVEAN predicted 10, PANTHER-PSEP predicted 24, SNAP2 predicted 11, and PHD-SNPg predicted 30 nsSNPs (Fig. 1b; Supplementary Table S1) as deleterious, which were further used for structure-based consequence analysis of nsSNPs.
Structure-based prediction of functional consequences of nsSNPs
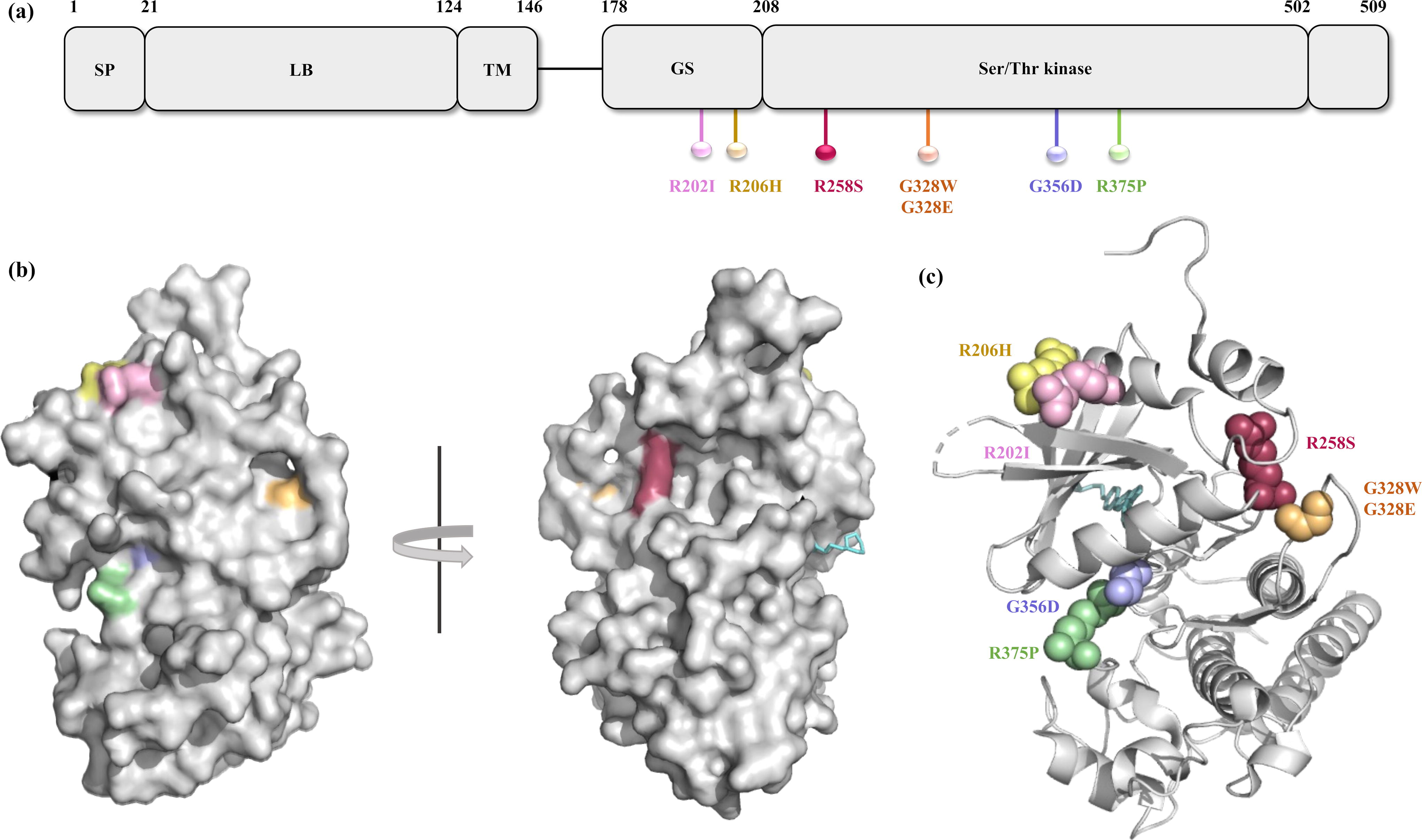
The seven predicted deleterious nsSNPs based on the sequence-based analysis were further evaluated with structure-based analysis tools—DUET, mCSM, SDM, DynaMut, INPD-3D, and CUPSAT. Variant R202I, R206H, R258S, G328E, G328W, G356D, and R375P were conclusively predicted to be deleterious based on the Gibbs free energy (ΔΔG kcal/mol) (Supplementary Table S2, Fig. S1d). Figure 2a illustrates the location of predicted deleterious nsSNPs across the domains of ACVR1 protein.
Evolutionary conservation analysis of ACVR1 gene
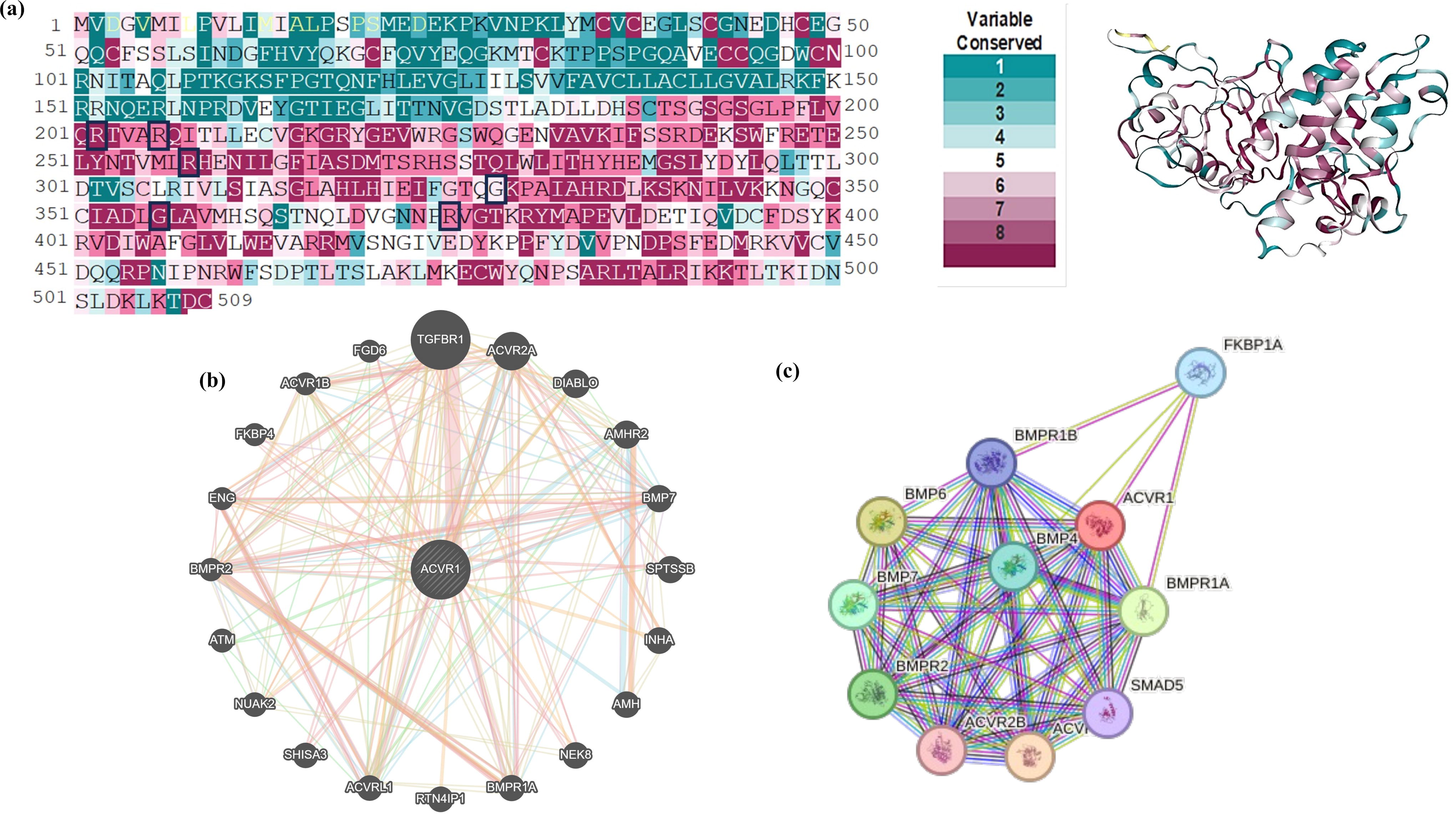
The evolutionary conservation profile was carried out for the ACVR1 protein sequence and structure. Mutation R202I, R206H, G356D, and R375P were predicted as functional and exposed residues with a score of 8, 9, 8, and 9, respectively, whereas G328E, G328W, and R258S were predicted as exposed residues with a score of 9, 6, and 6, respectively. Predicted deleterious mutations R202I and R206H lie in the GS domain, whereas R258S, G328E, G328W, R356D, and R375P lie in the Ser/Thr kinase domain, which was predicted as highly conserved domains (Figs. 2a and 3a).
Interaction network analysis
The gene–gene interaction network of ACVR1 within the cell was predicted by GeneMANIA (Fig. 3b). Physical interactions of ACVR2A, ACVR1B, ACVRL1, AMH, AMHR2, BMPR1A, BMPR2, BMP-7, ENG, FKBP4, FGD6, INHA, NEK8, NUAK2, SHISA3, SPTSSB, and TGFBR1 were predicted with ACVR1. Coexpression was predicted with ACVR2A, ACBRL1, AMH, AMHR2, BMPR1A, BMPR2, BMP-7, ENG, FKBP4, INHA, NUAK2, and TGFβR1. ACVR1 showed an interlink with ACVRL1, ACVR2A, ACVRL1, AMH, AMHR2, BMPRIA, BMPR2, BMP7, ENG, and TGFβR1. ACVRL1, BMP-7, EMG, and FGD6 were found to show colocalization with ACVR1. Genetic interactions of ACVRL1, ACVR1B, ACVR2A, AMHR2, ATM, BMPR2, BMP7, DIABLO, ENG, FGD6, NUAK2, SPTSSB, and TGFBR1 were predicted with ACVR1. The STRING database was utilized to construct a multinode interaction network of ACVR1 to identify and annotate all significantly important protein interactions amid cellular macromolecules. Network analysis showed 11 nodes, 46 edges, and 8.36 average node degrees. The average local clustering coefficient and the PPI enrichment value were 0.923 and 3.66e-15, respectively. Furthermore, PPI network of ACVR1 (Fig. 3c) revealed crucial interactions with BMP-2, BMP-4, BMP5, BMP-6, BMP-7, BMPR2, FKBP1A, SMAD1, SMAD5, and SMAD9.
Functional consequences of ACVR1 SNPs
Server HOPE was utilized to determine the consequences of the seven commonly predicted deleterious nsSNPs of ACVR1 from various tools (Supplementary Fig. S5a–g). It compares the changes in charge, size, and hydrophobicity of WT residues with the newly incorporated mutant residue. The alteration of the crucial residues impacts the domains of ACVR1. In R202I, the mutant residue isoleucine was smaller, more hydrophobic, and neutrally charged. In R206H, the histidine (neutral) mutant residue was smaller than the WT residue arginine (positive). The structural alteration showed that the substitution might result in a loss of interactions, abolishing protein functioning. In R258S, the mutant residue serine was smaller, neutrally charged, and more hydrophobic. The loss of positively charged WT residue may cause molecular interaction loss.
In the case of G328E and G328W, the mutant residues E and W were bigger, negatively charged, and hydrophobic compared with the WT residue. Being on the protein’s surface and most flexible residue, this mutation was found to be convincible to disorder the backbone and molecular interactions of the protein. The G328E mutation involves the substitution of glycine with glutamic acid, a larger negatively charged residue. This change can disrupt local electrostatic interactions and hydrogen bonding networks, potentially leading to moderate destabilization and altered kinase activity. In contrast, the G328W mutation replaces glycine with tryptophan, one of the largest amino acids with a bulky aromatic side chain. This substitution introduces significant steric hindrance, disrupting the hydrophobic core and likely causing misfolding of the kinase domain.
Consequently, the G328W mutation is expected to have a more severe impact, possibly abolishing the kinase’s ability to bind Adenosine triphosphate or substrates, thus completely impairing its enzymatic function. The distinct biochemical properties of these substitutions result in the following different cellular consequences: G328E may retain some residual activity, causing altered but functional signaling, whereas G328W is likely to lead to a more severe loss of function, profoundly affecting cellular pathways. In G356D, the mutant residue was negatively charged and, due to its bigger size, experienced hindrances in fitting to the protein core. The unusual torsion angle of the residue might disturb the local backbone conformation and cause a loss of flexibility. In R375P, the mutant residue was more minor, neutrally charged, and hydrophobic. The mutation disturbs the ionic interactions of the hydrogen bond formed by the WT residue arginine with serine at 244, aspartic acid at positions 336 and 354, and the salt bridge formed with aspartic acid at 241, glutamic acid at position 248, and aspartic acid at positions 336 and 354. Comprehensively, the mutation occurring in the residues of the GS domain usually impacts the functional binding of the protein; the hydrophobicity alteration of residues in the Ser/Thr kinase domain may result in the loss of hydrogen bonds, disturbed protein folding, and altered signal transduction; and the mutations in the protein kinase domain can disrupt the protein functioning.
Molecular dynamic simulation analysis
The molecular dynamic simulation of seven modeled mutants of ACVR1 (R202I, R206H, R258S, G328E, G328W, G356D, and R375P) was performed for around 400 ns to analyze their dynamic information concerning their reference WT ACVR1 protein structure (Fig. 4a to 4h and Supplementary Fig. S6a to S6g). The RMSD (root-mean-square deviation) curves for the C-alpha backbone atom were analyzed to predict the system’s overall stability during the simulation interval. The RMSD trajectory R202I was stable from 250 ns to 400 ns at 0.5 nm. In contrast, the RMSD trajectory for R206H and R258S was considerably stable from 50 to 200 ns at 0.4 nm and 200 to 200 ns at 0.4 nm; furthermore, an elevated shift was observed with stability from 300–400 ns at 0.45 nm, respectively. The G328E mutant exhibited stable RMSD from 100 to 250 ns at 0.4 nm and scaled upwards to 0.5 nm till the end of the simulation at 400 ns, whereas G328W stabilized from 280 ns at 0.42 nm till the end of the 400 ns simulation. Moreover, the mutant G356D showed a stable trajectory from 50 to 200 ns at 0.4 nm, and RMSD of mutant R375P swept up from 250 to 400 ns at 0.45 nm after being stable at 0.4 nm from 100 to 250 ns. Taken together, the MD simulation results verified that most of the mutations caused an increase in the RMSD value in the mutant systems relative to the WT system. Consequently, the mutations in the protein cause structural variations leading to functional disturbances.
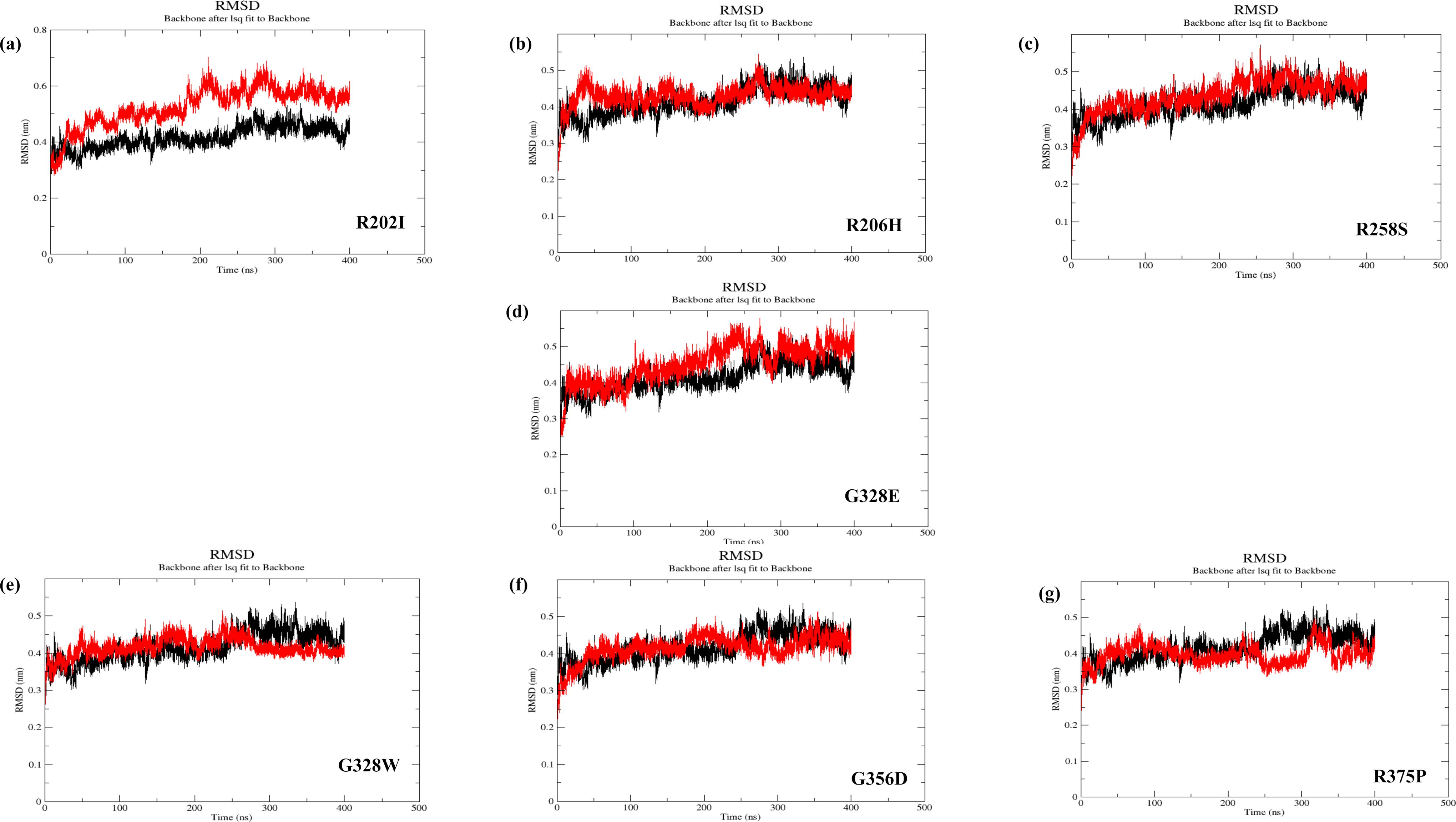
RMSD curve of C-alpha carbon for ACVR1 proteins;
Essential dynamics analysis
The EV projections display the essential dynamics that have been displayed in Supplementary Fig. S7a–h for the top 10 eigenvalues against the percentage of variance in the squared pose changes in the covariance matrix out of 990 EVs. The top 10 EVs explained most of the variance in the data, indicating the dominant motions of the ACVR1 protein. In all the mutated protein structures of the ACVR1 protein, including the reference, a decrease in variance percentage for the three eigenmodes (with over 75% coverage) was observed. Following the elbow angle shift, the drip for all the mutated structures and the reference indicated the structural stability for eigenvalues ranging from 4 to 10. Exceptionally, in mutated structures R202I and R258S, change in elbow position was observed at three eigenvalues.
Furthermore, a decrease in contributions from the eigenvalues suggested that the mutated structure exhibited stability. These findings align with our analysis of protein fluctuations caused by mutations through dynamic simulations. The limited fluctuations in the target protein structure may be due to mutations near its binding site. Out of 10 EVs, the reference protein explained the structural variability by the first EV as 46.76%. In contrast, the other mutation models explained the maximum variability coverage as 86.52% in R202I, 57.65% in R206H, 52.54% in R258S, 61.75% in G328E, 27.73% in G328W, 34.89% in G356D, and 41.79% in R375P. These observations conclude that restricting motion in the target protein due to mutation can impact its activity and potentially inhibit its function.
Free energy landscapes analysis
FELs analysis, based on Gibbs free energy, is crucial for determining whether the protein prefers an unfolded or folded state. In this analysis, higher Gibbs free energy (red spots) indicates an unfolded protein state, whereas lower Gibbs free energy (deep blue spots) represents a folded state. Figure 5a–h shows the FELs of WT and mutated structures. Deeper blue regions represent different conformational states, including global or local free energy minima. R202I and G356D, in particular, showed wider basins than the reference proteins, suggesting a more stabilized state in a metastable conformation. All ACVR1 mutations, except for G328E, exhibited darker blue patches than the reference protein, indicating the presence of several metastable states and protein destabilization.
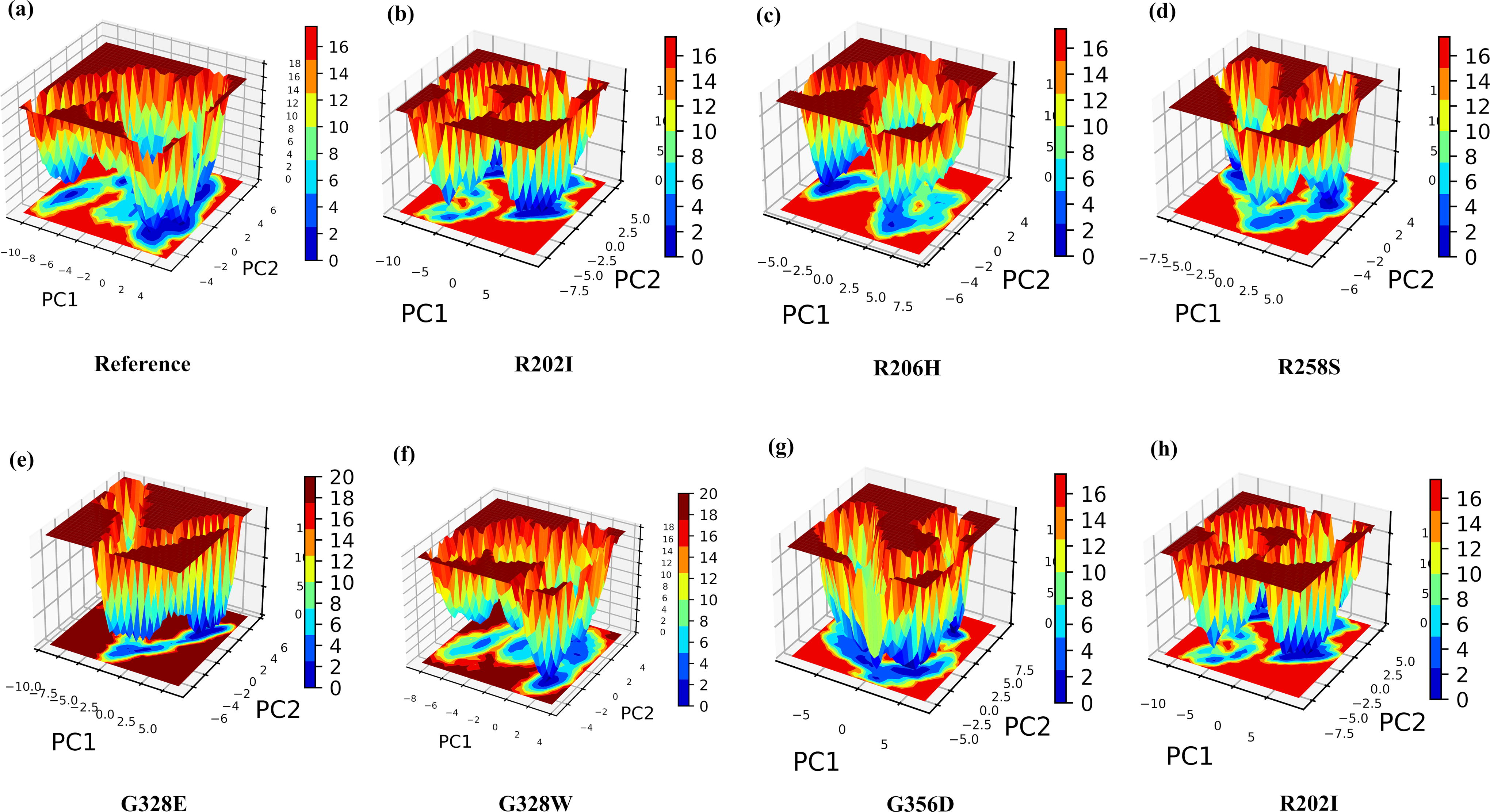
The Gibbs energy landscape captured during a 400 ns MD simulation for
Disease association of deleterious ACVR1 nsSNPs
The existing literature to date has identified the association of deleterious variants, viz. R206H, G328E, G328W, and G356D with FOP and DIPG, whereas R202I, R258S, and R375P were found to be uniquely associated with FOP (Han et al., 2018). Furthermore, the R206H and G328E mutations were identified to be associated with PFE (Pratt et al., 2022). The mutational profile on cBioPortal and canSAR web server reported the association of R206H with moderate severity in Pediatric Brain Cancer-Differential Expression (PBCA-DE) and Uterine Corpus Endometrial Carcinoma-Cancer Genome Atlas Program (UCEC) and G356D with uterine endometrioid carcinoma (Coker et al., 2019; Gao et al., 2013). However, neither the canSAR nor cBioPortal’s mutational profile of ACVR1 uncovered the significant association of other nsSNPs with any specific cancer.
Outlook and Conclusions
The variants of ACVR1 are foreseen to influence the binding affinity of Smad effector protein receptors and, in turn, their autophosphorylation. With direct functional and protein modeling analysis, others (Shore and Kaplan, 2011; Shore and Kaplan, 2010) have shown in previous studies that such gain-of-function mutations result in increased downstream transcriptional activity. Attributable to the significance of nsSNPs, the current in silico study investigates the impact of nsSNPs on the structural and functional integrity of ACVR1 protein. Several multitudinous bioinformatics tools were assessed, encompassing the commixture of ambiguous approaches for analyzing the nsSNPs. From the National Center for Biotechnology Information (NCBI) database among 5095 SNPs, 339 nsSNPs of the ACVR1 protein were screened from NCBI and taken forward for further in silico analysis. The consolidated sequence and structure-based bioinformatic strategies predicted R202I, R206H, R258S, G328E, G328W, G356D, and R375P as deleterious variants significantly impacting the structural and functional activity of the ACVR1. The STRING and GeneMANIA databases also revealed the interaction network that the mutations can influence.
Likewise, HOPE and ConSurf servers predicted these mutations’ physiochemical and evolutionary impact on the protein structure and its conserved residues. Certainly, no mutations have since been reported to occur in the ligand-binding domain. The identified deleterious mutations in ACVR1 protein are sighted in the GS activation domain or serine threonine kinase domain, ensuring a functional protein. The GS domain is an intracellular On/Off switch that harbors R202I and R206H mutations, whereas the serine threonine kinase domain accommodates the remaining deleterious mutations R238S, G328E, G328W, G356D, and R375P. The GS domain shows sequence conservation among type I receptors in TGFβ family members; hence, the mutation in such regions was predicted to have considerable influence on the structure and function of the protein. MD simulation, PCA, and FEL analysis were further used to determine the stability of WT and mutant protein structure. The disease association analysis revealed the association of deleterious SNPs with FOP, DIPG, PFE, PBCA-DE, and UCEC.
The aforementioned nsSNPs were recognized as possible risk factors due to their explicit impact on the structure and function of native proteins. However, the current study has some limitations, is inadequate, and needs more experimental verification to understand and elucidate the potential impact of deleterious nsSNPs on the ACVR1 protein. The computational tools based on different algorithms have confined boundaries limiting predictions and requiring functional studies for further in vitro validations.
The current in silico analysis has given a better insight into how the nsSNPs can significantly alter the structure of the protein. Uncovering the harmful variants and extended phenotype–genotype association studies may pave the way for future exploration of precision medicine. The predicted candidate mutations of ACVR1 are associated with some rare conditions; nonetheless, a gap still exists in the less explored field of cancer research for further validation.
Footnotes
Acknowledgments
G.N. sincerely thanks the Council of Scientific and Industrial Research (CSIR), Government of India, for providing research fellowship. The authors sincerely thank Dr. Shiv Bharadwaj for his constant support, advice, and valuable comments on postmolecular dynamic simulation analysis.
Data availability statement
All data pertinent to the study contents and conclusions are in the article and Supplementary information file. The authors can provide any further datasets and information upon reasonable request.
Authors’ Contributions
G.N.: Conceptualization, methodology, computational analysis, data visualization, and writing original draft. S.R.R.G.: MD simulation analysis and interpretation. V.R.: PCA, structure variability, FEL analysis, and interpretation. R.K.P.: Computational analysis, interpretation, and editing. M.P.: Computational analysis, interpretation, and language editing. A.S.: Conceptualization, reviewing, and editing. I.K.S. and M.P.: Conceptualization, methodology, computational analysis, reviewing, editing, and supervision. All authors contributed significantly and read, reviewed, and approved the final article.
Author Disclosure Statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Funding Information
I.K.S. is thankful to the
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.