
Abstract
Precision oncology promises individually tailored drugs and clinical care for patients with cancer: That is, “the right drug, for the right patient, at the right dose, and at the right time.” Although stratification of the risk for treatment resistance and toxicity is key to precision oncology, there are multiple ways in which such stratification can be achieved, for example, genetic, functional pathway based, among others. Moving toward precision oncology is sorely needed in the case of acute lymphoblastic leukemia (ALL) wherein adult patients display survival rates ranging from 30% to 70%. The present study reports on the pathway activity signature of adult B-ALL, with an eye to precision oncology. Transcriptome profiles from three different expression datasets, comprising 346 patients who were adolescents or adults with B-ALL, were harnessed to determine the activity of signaling pathways commonly disrupted in B-ALL. Pathway activity analyses revealed that Ph-like ALL closely resembles Ph-positive ALL. Although this was the case at the average pathway activity level, the pathway activity patterns in B-ALL differ from genetic subtypes. Importantly, clustering analysis revealed that five distinct clusters exist in B-ALL patients based on pathway activity, with each cluster displaying a unique pattern of pathway activation. Identifying pathway-based subtypes thus appears to be crucial, considering the inherent heterogeneity among patients with the same genetic subtype. In conclusion, a pathway-based stratification of the B-ALL could potentially allow for simultaneously targeting highly active pathways within each ALL subtype, and thus might open up new avenues of innovation for personalized/precision medicine in this cancer that continues to have poor prognosis in adult patients compared with the children.
Introduction
Acute lymphoblastic leukemia (ALL) is a veritable challenge in the oncology clinic, particularly for adult patients. Unlike the ALL in childhood, wherein the long-term survival rate approaches to 90% (Pui et al., 2015), adult patients have markedly less favorable treatment outcomes, with long-term overall survival rates ranging from 30% to 70% depending on the age group, subtype of the disease, and the treatment applied (Hayakawa et al., 2014; Kantarjian et al., 2004; Lennmyr et al., 2019; Rowe et al., 2005; Rytting et al., 2016; Stock et al., 2019; Stock et al., 2013). Precision oncology scholarship aims to unpack the mechanistic heterogeneity in cancers and thereby invites the clinicians to move toward individually tailored treatments in ALL that can offer greater safety and efficacy. However, the ways in which precision oncology should stratify patients with ALL to improve their treatment remain unclear. This calls for a deeper understanding of the ALL pathophysiology so as to identify the most promising avenues of therapeutics innovation for patients with ALL.
Insofar the pathophysiology is concerned, ALL arises from uncontrolled proliferation of abnormal cells, resulting in a blockage in the maturation process of lymphoid precursor cells. Chromosomal rearrangements and structural variations that disrupt lymphoid maturation, regulation of cell growth and proliferation, and epigenetic regulation are frequently observed in ALL (Mullighan, 2013). Interestingly, these chromosomal variations are accompanied by fewer somatic single nucleotide variations than other cancer types (Iacobucci and Mullighan, 2017). These variations frequently lead to changes in lymphoid development, cytokine receptors, kinases and Ras signaling pathways, pathways associated with tumor suppression, and chromatin modifications (Chiaretti et al., 2014).
ALLs are classified immunophenotypically into three major groups: B-cell, T-cell, and mixed phenotype ALL where myeloid and lymphoid expression can be observed. All three major groups are genetically and molecularly heterogeneous and have multiple subtypes. Genomic studies have paved the way for identifying recurrent genetic alterations in ALL, leading to characterization of new subtypes and facilitating the establishment of a detailed and layered classification (Iacobucci and Mullighan, 2017; Roberts, 2018).
A key reason for poorer outcomes in adult ALL stems from the current lack of precision medicines for high-risk subtypes that are more prevalent within this age group (Iacobucci and Mullighan 2017; Roberts, 2018). For example, the frequency of Ph-positive leukemia in adults was reported to be 34% of ALL, with a peak incidence at 50–59 years (Lennmyr et al., 2019), while this is 2% in pediatric ALL (Zhang et al., 2022). Another high-risk ALL, Ph-like ALL, accounts for about 10–15% of childhood ALL cases but increases to over 20% in adults, peaking at 25–30% in adolescents and young adults (Roberts, 2018; Zhang et al., 2022). Adults often have comorbidities and decreased tolerance to intensive chemotherapy regimens, which leads to lower treatment adherence and increased toxicity (Kantarjian et al., 2004; Siegel et al., 2018; Wann et al., 2021). In addition, adults have a higher prevalence of minimal residual disease after treatment, increasing the likelihood of relapse (Bassan and Hoelzer, 2011). While the adolescent and adult ALL cases benefit from treatment regimens used in pediatric ALL (Stock et al., 2019), specific subtypes of B-ALL, such as B-other and Ph-like ALL, remain high risk for which current treatment regimens are not sufficiently effective. B-other ALL represents a heterogeneous group with less understood genetic background, usually defined by the absence of all routinely evaluated classifying aberrations (Zaliova et al., 2019). Since the latter patients in the high-risk adult group cannot be detected with routine tests, they receive standard chemotherapy regimens, which is another factor negatively affecting treatment outcomes.
The molecular subtypes of ALL are typically identified through cytogenetic analysis, which detects specific translocations or through techniques that identify small-scale molecular variations. These recognized subtypes include hyperdiploidy, characterized by the nonrandom gain of at least five chromosomes; hypodiploidy, characterized by fewer than 44 chromosomes; and recurrent translocations. Examples of these translocations include ETV6:RUNX1 encoding t(12;21)(p13;q22), TCF3:PBX encoding t(1;19)(q23;p13), BCR:ABL1 (also known Ph-positive) encoding t(9;22)(q34;q11), and mixed-lineage leukemia (MLL) rearrangements involving 11q23 with various common partner genes. These alterations serve as reference points for the molecular subtypes of ALL.
Almost 70% of all B-ALL cases can be classified using cytogenetic analysis (Mrózek et al., 2009). Extensive sequence analyses detailed the genomic profile of B-ALL and led to the identification of new subtypes (Alaggio et al., 2022; Montaño et al., 2018). Some of the newly identified subtypes include genetic alterations, such as MED2D:ZNF384 rearrangement, any fusion pair involving the DUX4 gene, or alterations in the PAX5 and IKZF1 genes. Other subtypes are defined by changes in gene expression profiles such as “Ph-Like” ALL and “ETV6:RUNX1-like” rather than cytogenetically. However, the existence of cases that cannot be classified even with these comprehensive sequencing approaches and whose prognosis cannot be foreseen calls for new ways of thinking about how best to stratify patients and risks to achieve the overarching goal of precision oncology with improved clinical outcomes.
Global DNA/RNA sequencing analysis has shown that ALL subtypes are characterized by several cooperative oncogenic lesions that affect genes that have a crucial role in the proliferation and emergence of the leukemic cell clone. Next-generation sequencing technologies have recently identified many novel lesions in ALL and helped improve our understanding of its pathogenesis. These lesions play crucial roles in key signaling pathways associated with the pathogenesis of ALL (Bhojwani et al., 2015; Iacobucci and Mullighan, 2017; Mullighan, 2013). Similarly, many other genes become activated or inactivated because of the presence of specific genomic alterations or complex structural rearrangements, or are affected by copy-number changes (Van Vlierberghe et al., 2008). However, putting the large number of genomic alterations investigated in the functional context of the pathways they alter may provide additional and hitherto missing crucial information on mechanisms of pathogenesis and opportunities for individually-tailored treatments. Therefore, quantifying and inferring the disruption of signaling pathways remain a knowledge gap that needs to be addressed for an in-depth understanding of ALL pathogenesis, therapeutics, and clinical outcomes.
Notably, the genetic and epigenetic heterogeneity attendant to ALL can be collectively captured at the transcriptome level, thus enabling the characterization of pathway activity signatures. Pathway-based approaches offer a potential advantage in identifying optimal drug choices or tailoring treatment strategies based on individual pathway activity patterns. Multiple studies have pointed out that drugs exert their effects by affecting entire pathways rather than individual components (Wang et al., 2021). This view, together with the recognition that many complex diseases such as leukemia result from dysfunctions in multiple biological pathways rather than isolated genetic or molecular factors, offers an opportunity for an innovative approach to drug discovery and development (Vasaikar et al., 2016).
The aim of the present study was to characterize the pathway signatures of B-ALL using transcriptomic profiles of patients with different genetic/molecular subtypes of B-ALL and with an eye to inform precision medicine for ALL.
Materials and Methods
Different datasets encompassing adolescent and adult patients were combined while using several computational models to derive the activity of the signaling pathways that are frequently perturbed. The present study used publicly available datasets and did not require ethics committee approval. The study was conducted under the overall research ethics oversight of the author’s institution.
Construction and preprocessing of the initial dataset
The datasets that were used in this study are given in Figure 1a. These datasets comprised cases from different age groups and encompassed various subtypes of ALL. Pediatric ALL samples were excluded (ages <15) from the study as we aimed to work with adolescents and adults, and these groups display poorer and more heterogeneous clinical outcomes. Because the study focused on B-ALL subtypes, T-ALL samples were not included in the dataset.
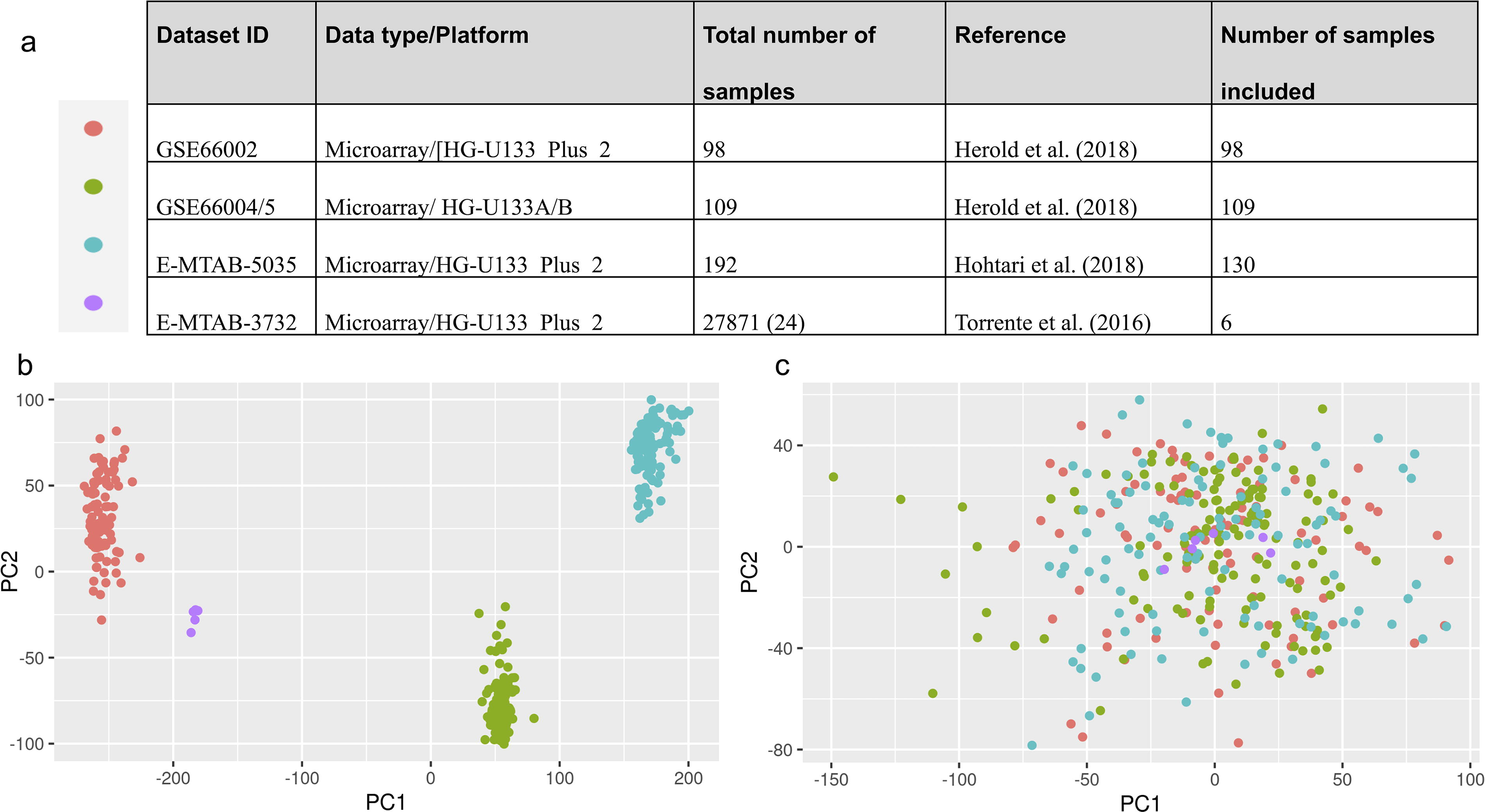
Distribution of datasets used in this study
The microarray data were normalized using the robust multiarray average (RMA) algorithm with the R package affy (Gautier et al., 2004), version 4.3.2. All data were filtered to include probe sets found on the Affymetrix U133A or U133B platforms. The U133A and U133B platforms complement each other, offering a comprehensive analysis of gene expression. The HGU133Plus2 platform includes all the probe sets from both the U133A and U133B platforms and more probe sets, providing broader coverage of the human transcriptome. Since the dataset contained B-ALL samples from different studies, it was necessary to consider technical variation (batch effect, etc.). We also included six normal precursor B-cell samples as a reference at this stage (Torrente et al., 2016). To eliminate technical differences, the Bayesian factor regression modeling method was applied, as described by Johnson et al. (2007) and implemented in vsa R package, version 3.48.0 (Leek et al., 2023). The extent to which technical variation was removed was visually examined using principal component analysis (Fig. 1b) using prcomb function of base R (version 4.3.2) package stats (R Core Team, 2023). Accordingly, the normalized and technically adjusted gene expression matrix was constructed for subsequent analyses.
Calculation of pathway activities from gene expression data
The Progeny R package (Schubert et al., 2018) uses HUGO Gene Nomenclature Committee (HGNC) gene symbols; therefore, Ensembl gene names were converted to HGNC gene symbols using the https://www.genenames.org/ website. As a result, a gene expression matrix containing 18,540 genes and 337B-ALL samples was generated. Standardized pathway activities were calculated for each patient using the generated gene expression matrix and the Progeny R package, version 1.22.0 (Schubert et al., 2018). For this purpose, the 1000 most sensitive genes were used for each pathway.
Clustering patient samples based on pathway activity profiles
The consensus clustering algorithm developed by Monti et al. (2003), implemented with the R package ConsensusClusterPlus, version 1.64.0 (Wilkerson & Hayes, 2010), was used to perform clustering of patients and determine the optimal number of clusters. The optimal number of clusters is the number that best represents the underlying data structure, providing clear and distinct groups. Two different algorithms (HC, hierarchical clustering and PAM, partition around medoids) on a correlation-based distance were used to demonstrate the reproducibility of the results. For HC, a Ward-type linkage was used. The agreement between the two algorithms was quantified using the Jaccard index.
Results
Ph-like ALL resembles Ph-positive ALL on average pathway activity
Figure 2 presents the heatmap of calculated pathway activity values along with subgroup information for the samples. Visual inspection revealed the similarity between pathway activity patterns of Ph-like samples and Ph-positive samples. We calculated average pathway activity values for each subgroup (see Fig. 2a) and examined correlations between subgroups to demonstrate these similarities (see Table 1). Upon analyzing the average pathway activity, we observed that VEGF, TNFa, NFkB, and TGFb pathways showed high activity levels in both Ph-positive and Ph-like subtypes. It is worth noting that the p53 pathway exhibited significantly higher activity in the Ph-like subtype (0.43) in comparison with the Ph-positive subtype (−0.27). For the MLL-positive subtype, the pathways Trail and estrogen showed a positive average activity value. In addition, the B-other subtype displayed slightly elevated pathway activity values for mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K), and estrogen. The correlation values indicated how much Ph-like ALL resembled Ph-positive ALL on a pathway basis (correlation value: 0.82) and how it differed from other ALL subgroups (correlation values: −0.90 and −0.47) in our analysis. It is noteworthy that Ph-like ALL, despite being historically classified as B-other, differed from the B-other subtype regarding average pathway activity, with a correlation coefficient of −0.9. Compared with other subtypes, B-other ALL had a higher correlation with the MLL-positive subtype (0.35).
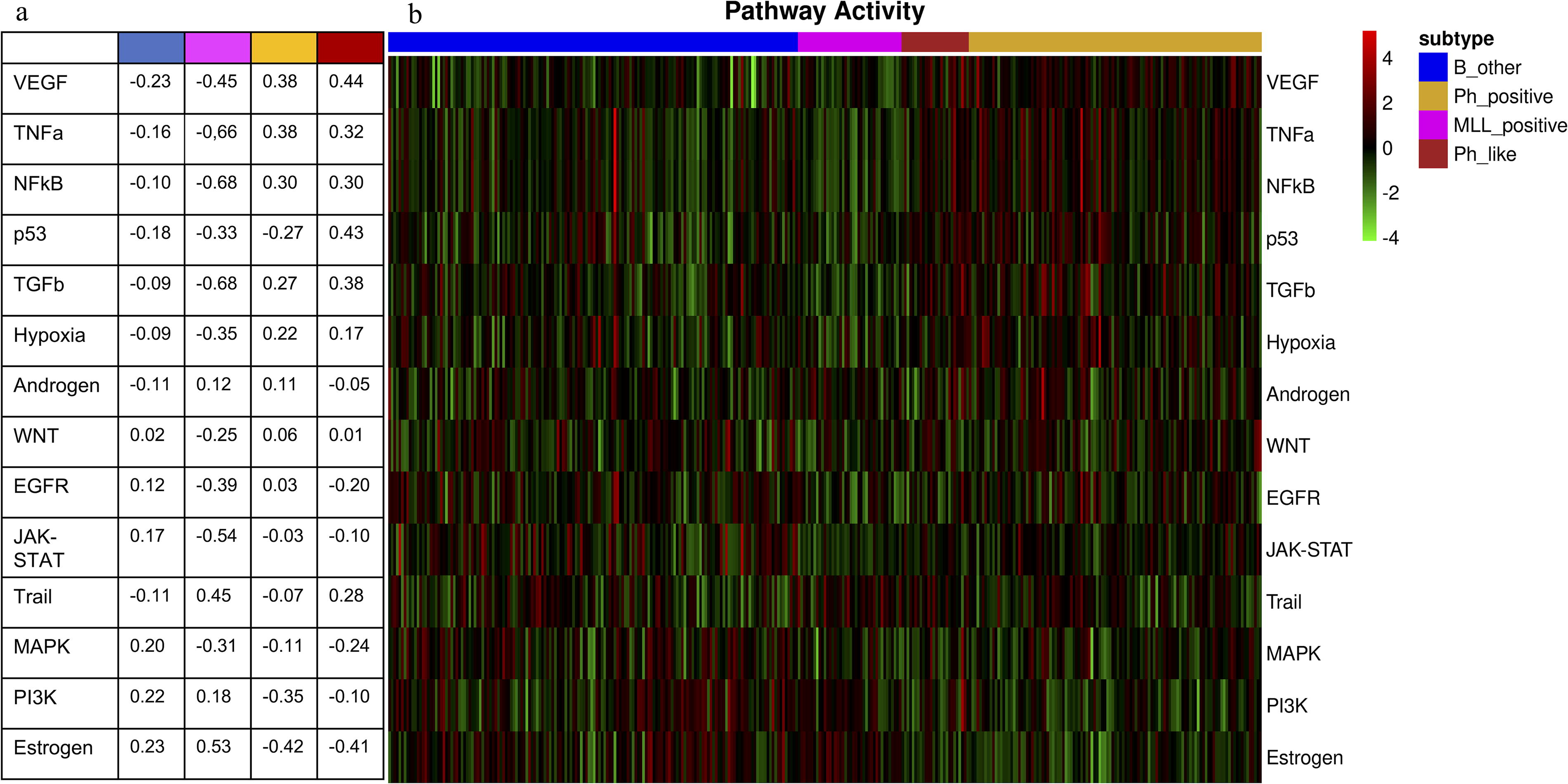
Average pathway activities
Correlation Values Between Average Pathway Activities Across B-ALL Subtypes
B-ALL, B cell acute lymphoblastic leukemia; MLL, mixed-lineage leukemia; Ph, Philadelphia chromosome.
B-ALL pathway activity patterns differ from genetic/molecular subtypes
Determining the optimal number of clusters in a given dataset is a challenging problem. However, using different unsupervised statistical learning techniques and seeking consensus between them is helpful in overcoming the problem of finding the optimal number of clusters. The consensus clustering approach provides quantitative evidence to the user for determining the optimal number of clusters and which cluster each patient belongs to. Determining the optimal number of clusters is possible by examining the consensus matrix, the consensus cumulative distribution function (CDF), and the relative change in the area under the CDF curve together. When the results of HC and PAM algorithms were evaluated together in terms of this evidence, we observed that the optimal number of clusters in the ALL dataset was 5. In addition to assessing the optimal number of clusters, we evaluated cluster composition by quantifying consensus among HC and PAM clusters using the Jaccard Index (see Table 2). Accordingly, we assessed the reproducibility of the clusters using different approaches and increased the reliability of our results. The Jaccard Indices obtained ranged from 0.45 to 0.77, indicating an adequate to high agreement between the clusters generated by HC and PAM (see Table 2).
The Agreement Between Clusters Generated by HC and PAM Quantified Using the Jaccard Index. The highest Jaccard Values, Indicating Corresponding Clusters, are Shown in Boldface
HC, hierarchical clustering; PAM, partition around medoids.
Figure 3 demonstrates how ALL subtypes are distributed into five clusters based on pathway activity patterns. Although we observed a tendency for some of the clusters (for instance, Cluster 3 overwhelmingly consisted of B-other samples, and Cluster 1 mostly consisted of Ph-positive samples), the distribution of ALL subtypes into clusters based on pathway activity patterns revealed a highly heterogeneous landscape (Fig. 3, Table 3). In Cluster 4, a high number of pathways were upregulated except for the pathways Trail, JAK-STAT, and Wingless-related Integration (WNT). Cluster 1, which mostly consisted of Ph-positive samples, stood out for the high activity level of the hypoxia, p53, EGFR, NFkB, and TNFa pathways. Cluster 3 consisted mainly of B-other samples and showed high PI3K, estrogen, and MAPK pathway activities. Cluster 2 had a clear high activity in only the Trail pathway. Although some samples in Cluster 5 had high activity in the VEGF pathway, Cluster 5 was prominent for high activity in the JAK-STAT and WNT pathways.
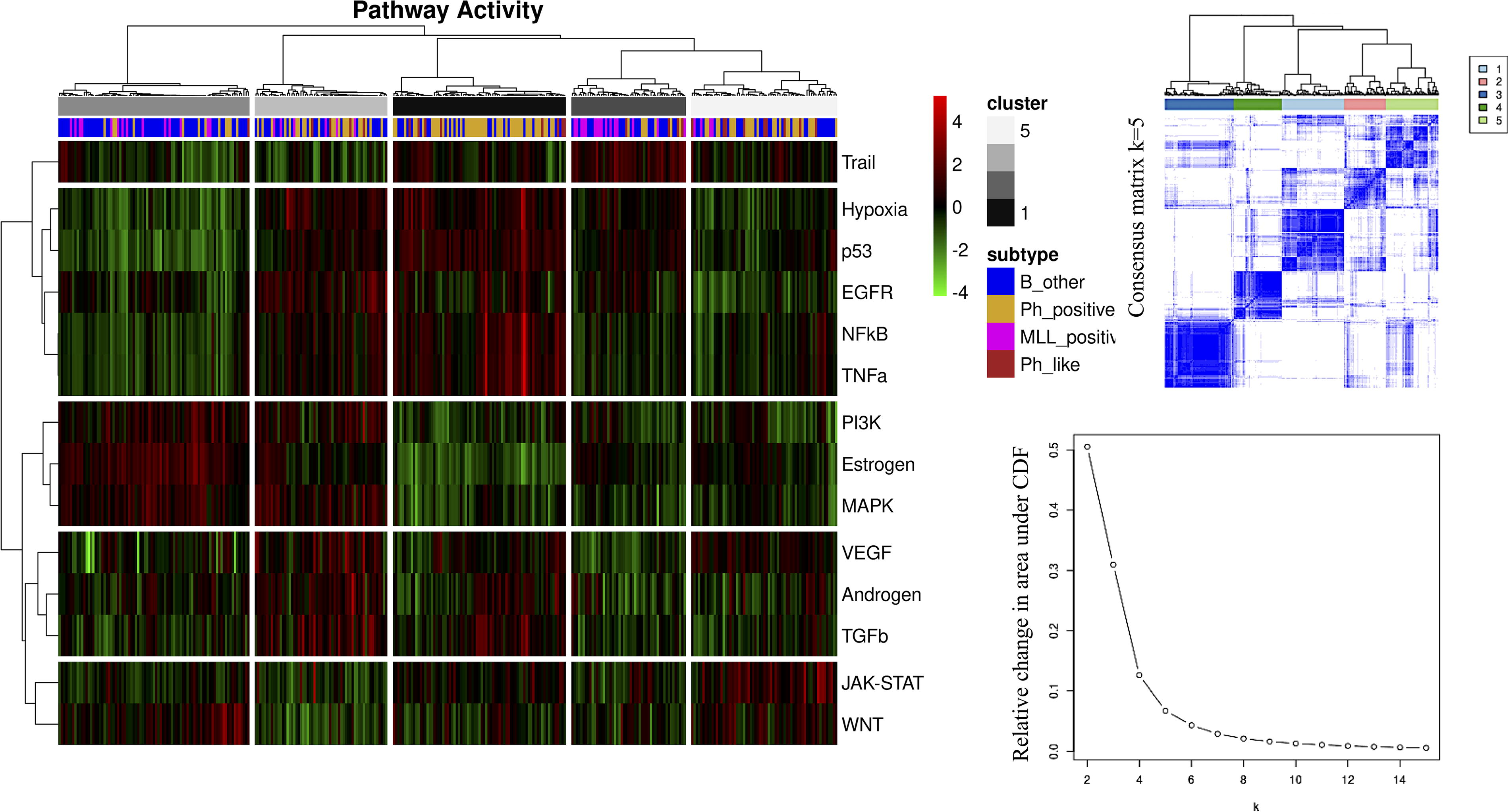
Consensus clustering and optimal number of clusters
Distribution of B-ALL Molecular Subtypes into 5 Different Clusters Based on Pathway Activity Profiles
B-ALL, B cell acute lymphoblastic leukemia; MLL, mixed-lineage leukemia.
The number in parentheses represents the percentage of samples belonging to the respective subtype within the corresponding cluster.
Discussion
The ALL pathogenesis and clinical outcomes are highly complex and heterogeneous. This calls for precision oncology and rational stratification of the disease and treatment risks and outcomes. On the contrary, the existing methods, such as genetic approaches to stratify ALL, have limitations, and new approaches to patient stratification and individually tailored treatments are much needed. In this context, the present study contributes to the literature by unpacking and highlighting the importance of pathway-based approaches in ALL to pave the way for future individually tailored therapeutics.
We combined different datasets and used multiple computational methods to reveal pathway signatures for adult B-ALL. We calculated the average pathway activity for each genetic/molecular subtype of B-ALL to provide a comprehensive overview. Considering the inherent heterogeneity within these subtypes, characterizing pathway-based subtypes appears to be considerably more appropriate and advantageous for the development of novel therapeutic strategies, such as combination therapies.
Ph-positive and Ph-like subtypes are classified as high risk and known for their unresponsiveness to current therapy regimens and, therefore, have inferior outcomes. The high average activity of pathways VEGF, TNFa, and NFkB is prominent for these subtypes and, hence, can be an explanation for inferior outcomes. Previous research has shown that leukemic cells secrete VEGF, which then interacts with receptors on the surface of endothelial cells. This interaction leads to the production of growth factors that affect the leukemic cells, causing them to increase their rate of proliferation and develop resistance to drugs (Song et al., 2012). High levels of tumor necrosis factor-alpha are associated with leukocytosis, high blast counts, and worse survival in patients with acute leukemia, indicating its involvement in the progression and relapse of the disease (Verma et al., 2022). Constitutive activation of the canonical NF-κB pathway, which blocks apoptosis and enhances cell proliferation to promote survival, was reported before for patients with ALL (Imbert and Peyron, 2017; Kordes et al., 2000).
Our findings reveal that the B-ALL pathway activity patterns diverge from genetic/molecular subtypes, which has important implications for personalized approaches in ALL treatment. It is now widely accepted that the root causes of many complex diseases stem from the dysregulation of multiple biological pathways rather than isolated genetic or molecular factors (Otero-Carrasco et al., 2024). Our findings once again confirm that this is also true for B-ALL. Although the scope of our work was limited to important signaling pathways, we demonstrated that the pathway activity patterns in B-ALL reveal a highly heterogeneous landscape, offering opportunities for combination therapies.
Cluster analysis identified five distinct clusters in adulthood B-ALL based on the pathway activity profile of patients. Each cluster possesses a unique pathway activity signature, which may have important implications for prognosis and treatment response. For example, Cluster 4, consisting of mostly B-other and Ph-positive cases that pose a challenge for traditional therapy approaches (Roberts, 2018), simultaneously exhibits the upregulation of multiple signaling pathways. The poor prognosis in these subtypes might be related to the number of dysregulated signaling pathways. Cluster 1, predominantly comprised of Ph-positive samples, displays increased activity in the hypoxia, p53, EGFR, NFkB, and TNFa pathways. Understanding the increased activity of these pathways may lead to targeted therapeutic approaches tailored to this cluster.
The Trail/TrailR system plays a vital role in regulating various biological responses in both tumor and normal cells. This involves triggering cell death through apoptosis and initiating nonapoptotic cell death signaling pathways (Montinaro and Walczak, 2023). Cluster 2 displays distinctively high activity in the Trail pathway, which is a favorable characteristic to overcome cancer given the role of this pathway in the cell and the efforts for the development of effective Trail activators (Montinaro and Walczak, 2023).
In Cluster 3, primarily consisting of B-other samples, increased activity was found in PI3K, estrogen, and MAPK pathways. Increased activity in the JAK-STAT and WNT pathways in Cluster 5 suggests that JAK-STAT and WNT inhibitors may benefit these patients. A previous study reported that among the potentially targetable alterations, JAK/STAT-class and RAS/RAF/MAPK-class aberrations were identified in 21% and 43% of B-other ALL patients (Zaliova et al., 2019), aligning with our current results.
Our results underscore the importance of understanding pathway activity patterns in B-ALL subtypes. Simultaneously targeting highly active pathways identified within each cluster may offer a personalized therapy approach, subsequently improving treatment outcomes and minimizing adverse effects. Further research addressing the pathway dysregulation in adulthood B-ALL cases is necessary to advance precision medicine approaches in leukemia treatment.
Conclusions
Despite advancements in treatment strategies, certain B-ALL subtypes such as B-other, Ph-positive, and Ph-like ALL remain high-risk categories with poor response to current treatment regimens. These high-risk subtypes are more prevalent in adolescents and adults, leading to poorer treatment outcomes. Importantly, this research underlines that ALL stems from disruptions in numerous pathways rather than a single genetic or molecular alteration, and that the pathway activity patterns in B-ALL diverge from genetic/molecular subtypes. Given the intrinsic heterogeneity among the patients of the same genetic/molecular subtypes, characterizing pathway-based subtypes seems notably more suitable and advantageous for advancing novel therapeutic strategies and drug development.
In all, a pathway-based stratification of the B-ALL could potentially allow for simultaneously targeting highly active pathways within each ALL subtype, and thus might pave the way for personalized/precision medicine in this cancer that continues to have poor prognosis in adult patients compared with the children.
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
The author gratefully acknowledges the financial support provided by Istanbul Bilgi University for the research conducted in this study (AK 85 080 0000).