
Abstract
Hurler–Scheie syndrome (MPS IH/S), also known as mucopolysaccharidosis type I-H/S (MPS IH/S), is a lysosomal storage disorder caused by deficiency of the enzyme alpha-L-iduronidase (IDUA) leading to the accumulation of glycosaminoglycans (GAGs) in various tissues, resulting in a wide range of symptoms affecting different organ systems. Postgenomic omics technologies offer the promise to understand the changes in proteome, phosphoproteome, and phosphorylation-based signaling in MPS IH/S. Accordingly, we report here a large dataset and the proteomic and phosphoproteomic analyses of fibroblasts derived from patients with MPS IH/S (n = 8) and healthy individuals (n = 8). We found that protein levels of key lysosomal enzymes such as cathepsin D, prosaposin, arylsulfatases (arylsulfatase A and arylsulfatase B), and IDUA were downregulated. We identified 16,693 unique phosphopeptides, corresponding to 4,605 proteins, in patients with MPS IH/S. We found that proteins related to the cell cycle, mitotic spindle assembly, apoptosis, and cytoskeletal organization were differentially phosphorylated in MPS IH/S. We identified 12 kinases that were differentially phosphorylated, including hyperphosphorylation of cyclin-dependent kinases 1 and 2, hypophosphorylation of myosin light chain kinase, and calcium/calmodulin-dependent protein kinases. Taken together, the findings of the present study indicate significant alterations in proteins involved in cytoskeletal changes, cellular dysfunction, and apoptosis. These new observations significantly contribute to the current understanding of the pathophysiology of MPS IH/S specifically, and the molecular mechanisms involved in the storage of GAGs in MPS more generally. Further translational clinical omics studies are called for to pave the way for diagnostics and therapeutics innovation for patients with MPS IH/S.
Introduction
Mucopolysaccharidoses (MPS) are a group of lysosomal storage disorders characterized by the accumulation of glycosaminoglycans (GAGs) owing to deficient lysosomal enzymes, leading to a myriad of cellular dysfunctions. Mucopolysaccharidosis type I (MPS I), an autosomal recessive disorder, arises from a deficiency in alpha-L-iduronidase (IDUA), an enzyme located in lysosomes that plays a crucial role in the degradation of dermatan sulfate and heparan sulfate (Clarke, 1993; Neufeld and Muenzer, 2001).
Based on the clinical phenotype, MPS I is further classified into three subtypes: Hurler syndrome (MPS IH; MIM: 607014), Scheie syndrome (MPS IS; MIM: 607016), and Hurler–Scheie syndrome (MPS IH/S; MIM: 607015). The estimated global prevalence of MPS I ranges from 2.48 to 7.1 per 100,000 population (Borges et al., 2020). The prevalence of MPS IH/S is estimated to be one in 115,000 (Marsden, 2017). For example, in India, MPS I represents over 25% of all cases of MPS, while MPS IH/S accounts for 10% of all diagnosed cases (Celik et al., 2021; Sheth et al., 2014). MPS IH is the most severe form and MPS IS is the most attenuated form of MPS I (Giugliani et al., 2010). MPS IH/S is an intermediate form of phenotypic expression (Muenzer, 2004). There is a considerable overlap of clinical manifestations among the three clinical syndromes, differing in age of onset, severity, and rate of progression.
However, the above phenotypic classification is neither precisely defined nor clinically distinguishable by analysis of enzyme activity or urinary GAG excretion (Oussoren et al., 2013). Thus, MPS I is clinically classified as severe (Hurler) or attenuated (non-Hurler), which includes MPS IH/S and MPS IS (Vijay and Wraith, 2005). The onset of clinical presentation ranges from shortly after birth, with rapid progression, to late onset and slow progression (Pastores et al., 2007). The most common clinical manifestation of MPS I include coarse facial features, progressive cognitive and neurological impairment, hearing loss, recurrent respiratory infections, corneal clouding, thickening of heart valves, hepatosplenomegaly, abdominal hernias, dysostosis multiplex, joint contractures, limited joint stability, and spinal stenosis (Alsafadi et al., 2021; Giugliani et al., 2010; Hampe et al., 2020).
In MPS IH/S, the phenotype presents in infancy with intermediate severity and may have mild or no cognitive impairment, but with variable clinical symptoms (Pastores et al., 2007). In MPS IS, patients present with symptoms later in childhood, progress less rapidly, and survive into late adulthood with preserved cognition function (Machnikowska-Sokolowska et al., 2023; Wraith and Jones, 2014). These constellations of progressive symptoms involving multiple organs are due to the abnormal storage of partially degraded GAGs due to the underlying IDUA deficiency (Swaroop et al., 2018).
GAGs are linear heteropolysaccharide chains involved in various biological processes (Zappe et al., 2022). GAGs are essential for maintaining structural integrity and hydration of the extracellular matrix (ECM), which is involved in cell signaling and cell-to-cell communication, and play active roles in cell adhesion, regulation of enzyme activity involved in inflammation, tissue remodeling, and pathogen interaction and defense (Aquino et al., 2010; Raman et al., 2005; Wang and Chi, 2022). In MPS, partially degraded GAGs accumulate in the lysosomes and ECM, which eventually causes dysfunction at the cellular and tissue levels in different organs of the body (Leal et al., 2022). In MPS IH/S, dermatan sulfate and heparan sulfate accumulate in the lysosomes (Clarke, 1993).
Earlier studies have attributed the clinical presentation solely to the accumulation of GAGs in organs and tissues (Wraith, 2013). However, subsequent experimental studies have observed that secondary and tertiary biochemical alterations, along with cellular changes, can significantly contribute to MPS pathology (Fecarotta et al., 2020; Tomatsu et al., 2018). Proteomic analysis of brain extracts from animal models of MPS I revealed abnormalities in metabolic pathways, neurotransmission, and cytoskeletal alterations, underscoring their contribution to the underlying neuropathology of this disease (Ou et al., 2017). Recent studies have analyzed the urinary proteome of patients with MPS to identify noninvasive biomarkers specific to each type of MPS. On the other hand, there was considerable discordance in the results obtained from these studies (Heywood et al., 2015; Yuan et al., 2019). The high degree of variability in protein abundance in urine may be attributed to factors such as dilution, contamination, hydration status of the patient, and sampling time (Joshi et al., 2024).
Previous studies have used animal models as well as urine and fibroblasts from patients with different types of MPS with the aim of identifying biomarkers to differentiate the different types of MPS (Baldo et al., 2015; Heywood et al., 2015; Liu et al., 2024; Ou et al., 2017; Randall et al., 2006). However, in most of these studies, they have focused on the severe form of MPS I (Hurler), and very few studies have focused on the proteomic changes in the attenuated (non-Hurler) subtypes of MPS I (Heppner et al., 2015; Liu et al., 2024; Ou et al., 2017; Zhang et al., 2022). In the present study, we investigated the cellular proteomic changes occurring in the moderate phenotype, that is, MPS IH/S. As there are reports of cellular dysfunction secondary to GAG accumulation, we also aimed to study phosphoproteomic alterations in MPS IH/S. Phosphoproteomics is a powerful tool for gaining deeper insights into altered signaling pathways and cellular dysfunction in a disorder (Rudolph et al., 2016). Few studies have explored the role of protein phosphorylation in MPS.
Postgenomic omics technologies offer substantial promise so as to better understand the changes in proteome, phosphoproteome, and phosphorylation-based signaling in MPS IH/S. Accordingly, we report here a large dataset and the proteomic and phosphoproteomic analyses of fibroblasts derived from patients with MPS IH/S and healthy individuals.
Materials and Methods
Cell lines
Fibroblasts from controls (n = 8) and MPS IH/S patients (n = 8) (Supplementary Table S1) were obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research in New Jersey, USA. The institutional review board at Mayo Clinic granted approval for this research (IRB19-004317).
Cell culture
Fibroblasts were cultured in minimum essential medium (MEM) alpha medium (Thermo Fisher Scientific, Gibco 12571) supplemented with 1% nonessential amino acids (Sigma Aldrich, M7145) and 15% fetal bovine serum (Thermo Fisher Scientific, Gibco A56707). The cells were then incubated in a CO2 incubator containing 5% CO2. After the cells reached 85–90% confluence, they were cultured for 12 h in serum-free MEM alpha medium without phenol red (Thermo Fisher Scientific, Gibco 41061). Following a 12-h incubation period, the cells were washed with phosphate-buffered saline (Thermo Fisher Scientific, Gibco 10010031). Cells were harvested using a 1X modified radioimmunoprecipitation assay buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.25% sodium deoxycholate, and 1 mM sodium orthovanadate) without sodium dodecyl sulfate. The harvested cells were stored at −80°C till further analysis. The experimental workflow employed in this study is illustrated in Figure 1.

Schematic workflow for phosphoproteomic and proteomic analyses of Hurler–Scheie syndrome (MPS I H/S) fibroblasts. Cells from MPS IH/S (n = 8) and control (n = 8) fibroblasts were lysed, and protein samples were digested with trypsin followed by labeling with tandem mass tags (TMT). Samples were pooled after bRPLC fractionation and phosphopeptides were enriched by IMAC using an automated AssayMAP BRAVO liquid handling system. The resulting samples (both bRPLC and IMAC fractions) were analyzed on an Orbitrap Fusion Lumos mass spectrometer. bRPLC, basic reverse phase liquid chromatography; IMAC, immobilized metal affinity chromatography.
Cell lysis, protein digestion, and TMT labeling
The cells were lysed using a probe sonicator (Branson SonifierSFX150) with an amplitude of 40% for a duration of 10 s for three cycles spaced every 5 min on ice. After centrifugation of the lysates at 10,000×g for 10 min at 4°C, the supernatant was used to estimate the protein concentration using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific,23227). Approximately 200 µg of protein was aliquoted from each sample for further analysis. The samples were treated with dithiothreitol (Sigma Aldrich, D9163-25G) at a final concentration of 10 mM and incubated at 37°C on a thermomixer with gentle agitation (300 rpm) for 45 min. Subsequently, iodoacetamide (Sigma Aldrich, I1149-100G) was added at a final concentration of 40 mM to the cooled sample, which was left in the dark at room temperature for 15 min. This was followed by protein precipitation using 5 volumes of ice-cold acetone. The sample was incubated at −20°C for 2 h, followed by centrifugation at 14,000 × g for 30 min.
The pellet was dissolved in 50 µL of 8M urea in 50 mM triethylammonium bicarbonate buffer (TEAB, pH 8.5). The samples were further diluted 10 times with 50 mM TEAB buffer (pH 8.5). TPCK-treated trypsin (Worthington, LS003744) was added at a final amount of 1:20 (trypsin:total protein). The mixture was incubated overnight at 37°C with mild shaking (750 rpm). The following day, the peptide digests were desalted using TopTipC18 tips (Glygen, TT2C18.96). After cleanup, peptide estimation was done using Pierce Colorimetric Peptide Assay kit (Thermo Fisher Scientific, 23275). An equal peptide amount from each sample (200 µg) was taken further for labeling with TMTPro16-plex (Thermo Fisher Scientific, A44520) as per the manufacturer’s protocol. Briefly, TMT regents resuspended in anhydrous acetonitrile were added to peptide samples resuspended in 100 mM triethyl ammonium bicarbonate (TEAB) buffer in the ratio of 2:1 (TMT reagent:peptide, wt:wt) and incubated at room temperature for 1 h. After incubation, the labeling reaction was quenched using 5% hydroxylamine solution and the samples were pooled and dried for subsequent steps.
Basic reversed-phase liquid chromatography and phosphopeptide enrichment
TMT-labeled peptides were fractionated by high-pH reversed-phase liquid chromatography using Dionex Ultimate 3000 (Thermo Fisher Scientific). Pooled peptides (∼3.2 mg) were resuspended in 5 mM aqueous ammonium formate at pH 9 (pH adjusted using ammonium hydroxide, Fisher Chemical, A669-500) and separated using a gradient of mobile phases A and B, on a 4.6 mm × 50 cm × 3.5 μm Xbridge column (Waters). Mobile phase A was composed of 20 mM ammonium formate in water, and mobile phase B was composed of 20 mM ammonium formate in 80% acetonitrile. Separation was carried out over a 2 h gradient from 2% to 40% mobile phase B. A total of 96 fractions were collected and concatenated into 12 fractions. A 20-μg equivalent of each fraction as calculated based on the initial input of 3.2 mg of peptides separated into 12 concatenated fractions was set aside for global proteome analysis, and the rest of the sample was dried down and stored at −80°C until phosphopeptide enrichment.
Phosphopeptide enrichment
Immobilized metal affinity chromatography (IMAC)-based phosphopeptide enrichment was performed using Fe(III)-NTA resin (Agilent Technologies, AssayMAP, G5496-60082) on an AssayMAP Bravo platform (Agilent Technologies). Briefly, Fe(III)-NTA cartridges were primed with 100 μL of 0.1% trifluoroacetic acid (TFA) in acetonitrile followed by equilibration with 100 μL of loading buffer (80% acetonitrile in 0.1% TFA). Samples were dissolved in 100 μL of loading buffer and loaded onto the cartridge. The columns were then washed with 50 μL of loading buffer, followed by phosphopeptide elution with 50 μL of 5% ammonium hydroxide/50% acetonitrile directly into 15 μL of 10% formic acid. Samples were dried down and stored at −80°C until subjected to LC–MS/MS.
LC–MS/MS analysis
LC–MS/MS analysis of both total proteome and enriched phosphorylated peptides was carried out on an Orbitrap Eclipse mass spectrometer (Thermo Scientific, San Jose, CA, USA) interfaced with a reversed-phase liquid chromatography system (Vanquish Neo, Thermo Fisher Scientific) as described earlier with modifications (Renuse et al., 2021). Briefly, the peptides were loaded onto a trap column (PepMapC18, 1.5 cm × 200 µm and 100 Å) using 0.1% formic acid at a flow rate of 10 µL/min. Subsequently, they were separated on an analytical column (PepSep 50 cm × 75 µm, C18 and 100 Å, Bruker) at a flow rate of 250 nL/min. Solvent B (100% acetonitrile and 0.1% formic acid) was set on a linear gradient of 3–50% over a period of 120 min, 50–95% of solvent B from 120 to 140 min followed by equilibration from 140 to 150 min.
Both the precursor and fragment ions were captured using an Orbitrap mass analyzer. With a resolution of 120,000 at 200 m/z, precursor ions in the m/z range 350–1800 were acquired. A higher-energy collisional dissociation method was employed to fragment the precursors with a normalized collision energy of 34. At 200 m/z, fragment ions were obtained at a resolution of 30,000. The scans were arranged with a 3 s cycle time between MS and MS/MS using the top-speed method. The capillary voltage for the ion transfer was maintained at 2.3 kV. A polysiloxane ion (m/z 445.120025) extracted from ambient air was used to activate the lock mass function for internal mass calibration.
Mass spectrometry data analysis
The data obtained after mass spectrometry analysis were searched against the Human RefSeq protein database (Version 81) using the SEQUEST and Mascot search algorithms on the Proteome Discoverer platform [version 2.1, (Thermo Fisher Scientific)]. We manually included proteins such as trypsin from porcine and bovine origin (UniProt entries: P00761, P00760, and Q29463), bovine serum albumin (UniProt entry: P02769) in the contaminants list. The search parameters on the Proteome Discoverer analysis composed of trypsin as the cleavage enzyme, two missed cleavages allowed, a precursor tolerance of 10 ppm, and 0.02 Da on the fragment ions. The fixed modification included carbamidomethylation at cysteine, TMTpro 16-plex (+304.2071 Da) modification at the N-terminus of the peptide, and lysine with variable modifications such as oxidation of methionine and deamidation of asparagine and glutamine. Dynamic modifications chosen for phosphoproteomic analysis included the oxidation of methionine; phosphorylation of serine, threonine, and tyrosine; and deamidation of asparagine and glutamine. The data were filtered with a 1% false discovery rate (FDR) after being compared to a decoy database.
To determine which proteins were differentially expressed or phosphorylated in comparison with the control group, and MPS IH/S data were analyzed using Perseus 2.0.7.0. software suite (Tyanova et al., 2016). The abundances were transformed to log2 scale and the normalization step was carried out based on width adjustment. The remaining missing values were then replaced with values based on normal distribution. Student t-test was performed and permutation-based FDR of 5% was applied to identify the differentially enriched and significant proteins. Further, multiple t-test ANOVA was performed with Benjamini–Hochberg test at FDR of 5%. Significant changes were identified by p < 0.05 and a fold change greater than twofold for proteins and phosphopeptides.
Bioinformatics analysis
Functional enrichment analysis for biological processes, molecular functions, and cellular components was performed using gProfiler (version e111_eg58_p18_30541362) with the g:SCS multiple testing correction method at a threshold of p-value <0.05 (Kolberg et al., 2023). Pathway enrichment was performed using ClueGO, a Cytoscape plug-in (Bindea et al., 2009). A network of protein–protein interactions (PPIs) was constructed using STRING (v.11.5) and was visualized using Gephi (v.0.9.7) software. Phosphorylation-specific dysregulated proteins were used in the construction of the interaction network (https://string-db.org/). A kinome map was built using the KinMap tool (http://www.kinhub.org/kinmap/index.html). Kinase-substrate enrichment analysis (KSEA) was performed using the online KSEA tool (https://casecpb.shinyapps.io/ksea/). Differentially phosphorylated proteins in the MPS IH/S group were analyzed using PhosphoSite Plus and NetworKIN as background datasets. The NetworKIN score cutoff, p-value cutoff (for plot), and number of substrate cut-offs were set to 2, 0.05, and 2, respectively.
Data availability
The mass spectrometry data obtained in this study have been submitted to the ProteomeXchange Consortium (Deutsch et al., 2020) via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD054788.
Results and Discussion
MPS IH/S is a subtype of MPS-I that occurs due to dysfunction of the gene IDUA, coding for IDUA, which results in impaired degradation of GAGs(Clarke, 1993). The degradation process occurs primarily in the lysosomes. When degradation is impeded, there is an increased accumulation of undigested GAGs in lysosomes, with subsequent spillage into the circulation. This accumulation eventually affects multiple organ systems in the body, resulting in the development of distinct clinical features depending on the type of GAGs being deposited (Fecarotta et al., 2020). The pathophysiology of MPS involves a complex cascade of events that encompass several subcellular processes and pathways. Considering this complex etiology, we aimed to investigate the proteomic and phosphoproteomic landscape of MPS IH/S to elucidate the molecular mechanisms underlying disease pathophysiology.
Quantitative proteomic profile of MPS I HS
The proteomic analysis yielded a total of 6,111 proteins, of which 17 proteins were upregulated [average log2 fold-change (MPS IH/S vs. Controls) ≥1; p < 0.05] and 14 were downregulated [average log2 fold-change (MPS IH/S vs. Controls) ≤−1; p < 0.05] (Supplementary Table S2). IDUA was observed to be downregulated (log2 fold-change = −1.165; p < 0.05), which aligns with the previously reported decrease in IDUA enzyme activity in these fibroblasts, as well as the clinical biochemical assays (submitted by the supplier to Coriell Institute for Medical Research) (Bach et al., 1993; Tieu et al., 1995; Tuyaa-Boustugue et al., 2023) (Fig. 2A, Supplementary Fig. S1A).
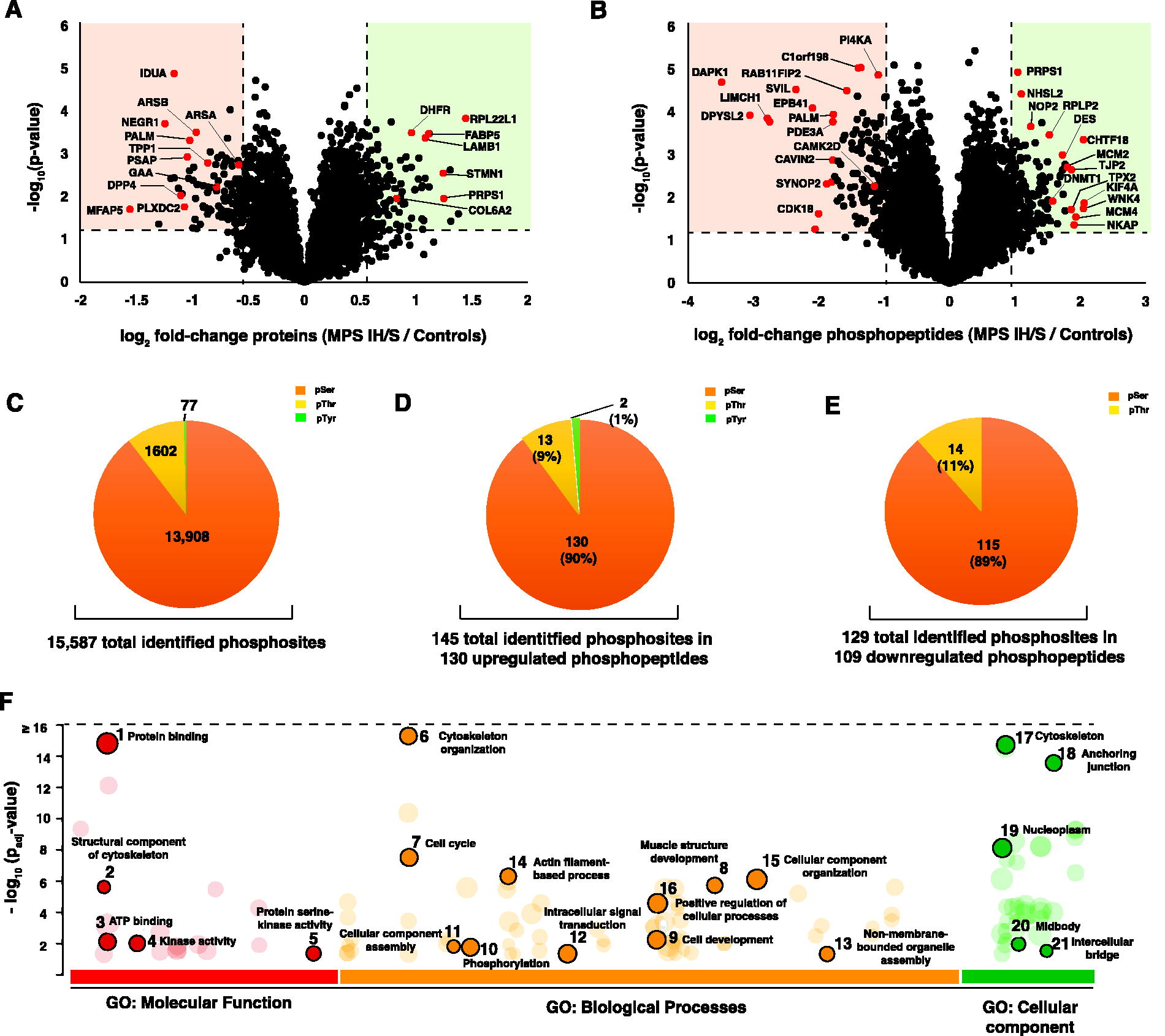
Proteomic and phosphoproteomic changes in Hurler–Scheie syndrome (MPS IH/S)
In addition to IDUA, other lysosomal enzymes such as cathepsin D (CTSD), prosaposin (PSAP), transglutaminase 2 (TGM2), and dipeptidyl peptidase-4 (DPP4) were downregulated, indicating that lysosomal function is affected beyond GAG degradation and contribute to impaired cellular function. Downregulation of DPP4 (average log2 fold-change = −1.10; p < 0.01) in fibroblasts might reflect the adaptive response to cellular stresses due to accumulation of GAGs, compared to upregulation of DPP4 observed in serum of MPS I H/S patients, reported by Liu et al., which might represent a compensatory response to the systemic inflammation or tissue damage observed in MPS IH/S (Liu et al., 2024).
Upon further analysis of the dataset, it was observed that key enzymes including arylsulfatase A, arylsulfatase B, acid alpha-glucosidase (GAA), and tripeptidyl peptidase 1 (TPP1) were downregulated which were above the set threshold log2fold change of −1 (average log2 fold-change ranging between −0.59 and −0.97; p < 0.05). The enzyme N-sulfoglucosaminesulfohydrolase (SGSH) (log2 fold-change = −0.70; p = 0.0012), which is directly involved in the degradation of heparan sulfate, is downregulated in association with impaired lysosomal function resulting from GAG accumulation (Bigger et al., 2018; Coutinho et al., 2012).
IDUA deficiency causes an increased accumulation of undigested heparan sulfate within lysosomes (Scott et al., 1991). The increased accumulation of heparan sulfate further inhibits other lysosomal enzymes, resulting in an impaired lysosomal environment that triggers the secondary accumulation of glycosphingolipids, phospholipids, cholesterol, and aggregation-prone proteins (Ago et al., 2024; Breiden and Sandhoff, 2020; Constantopoulos et al., 1980; Leal et al., 2022). Downregulation of key lysosomal enzymes such as CTSD, PSAP, and SGSH is probably in response to storage of GAGs resulting from IDUA deficiency, which initiates a cascade of secondary metabolic disturbances, adding to manifestations seen in MPS IH/S. The accumulation of heparan sulfate and other GAGs resulting from secondary metabolic disturbances may further exacerbate cellular stress and tissue damage, potentially leading to multi-organ involvement, including skeletal dysplasia and progressive systemic symptoms such as neurocognitive decline (Pierzynowska et al., 2023).
In addition to lysosomal enzymes, ECM and cell adhesion proteins (MFAP2, MFAP5, PLXDC2, and CAVIN2), signaling and receptor proteins (PALM, NEGR1, and SYNOP2), and metabolic enzymes (ADH1B and HNMT) were downregulated. Proteins involved in the cell cycle and DNA replication, such as thymidylate synthase (TYMS), stathmin 1 (STMN1), and kinetochore-associated protein (SPC24) were upregulated, suggesting increased cell proliferation, which is a probable mechanism to compensate the cellular stress or damage resulting from lysosomal dysfunction. This may explain the hyperplasia seen in certain tissues in MPS IH/S (Chu and Allegra, 1996; Kueh and Mitchison, 2009; McCleland et al., 2004).
Structural and microtubule-associated proteins (EML4, HIST2H2AB, and H1-1) were upregulated, possibly indicating an increased need for structural stability within the cell (Fry et al., 2016). ECM and cell adhesion proteins such as laminin subunit beta 1 (LAMB1) and podocalyxin-like protein (PODXL) are upregulated (Supplementary Figure S1B). This may be related to the deposition of dermatan sulfate, which can induce a wound-healing state in fibroblasts, thus indicating changes in the cellular microenvironment to repair the ECM, which might be disrupted in MPS (Bentley, 1967). The disruption in the ECM proteins occurring early in the disease history as observed in mouse models, could affect cartilage tissue structure and function, correlating with the connective tissue and skeletal abnormalities observed in MPS IH/S patients (Heppner et al., 2015).
Phosphoribosyl pyrophosphate synthetase 1 (PRPS1), which is involved in nucleotide biosynthesis, and enzymes involved in fatty acid metabolism (fatty acid binding protein 5 and SCD) were upregulated, suggesting metabolic alterations in response to cellular stress or increased biosynthetic activity in response to GAG accumulation. These alterations suggest the energy deficits and functional impairments seen in MPS IH/S patients (Heywood et al., 2015; Fu et al., 2021; Li et al., 2019). The proteomic analysis thus elucidates multiple biochemical pathways being affected in MPS IH/S, thereby providing critical insights in addressing the lysosomal dysfunction due to accumulation of GAGs and its downstream effects, which hold potential for development of therapeutic strategies aimed at managing the symptoms and improving the overall quality of life in patients affected with MPS IH/S.
Analysis of differential protein phosphorylation in MPS IH/S
Phosphoproteome analysis of fibroblasts from MPS IH/S patients identified 16,693 phosphopeptides derived from 4,605 proteins (Supplementary Table S3 and S4). A total of 15,587 phosphorylation sites were identified. Differential analysis yielded 147 hyperphosphorylated sites on 130 phosphopeptides from 116 proteins and 132 hypophosphorylated sites on 109 phosphopeptides from 82 proteins (log2 fold-change ≥1 or ≤−1, p < 0.05) in MPS IH/S compared to the controls (Fig. 2B, Supplementary Figure S2). The number and frequency of the identified amino acid phosphorylation sites was as follows: Ser-13,908 (89.2%), Thr-1602 (10.3%), and Tyr-77 (0.5%) (Fig. 2C, 2D, and 2E).
Functional categorization of phosphoproteins using gene ontology
Differentially abundant phosphopeptides were categorized using gene ontology (GO) annotation to understand the functional implications of MPS. These proteins are involved in cell cycle regulation, cytoskeletal organization, phosphorylation, and intracellular signal transduction. Phosphorylation has been detected in proteins associated with the cytoskeleton, anchoring junctions, nucleoplasms, and intercellular bridges. The enriched GO molecular function terms associated with differentially abundant phosphopeptides included protein binding, structural constituents of the cytoskeleton, ATP binding, kinase activity, and protein serine kinase activity (Fig. 2F; Supplementary Table S5). Most of these findings are in concordance with transcriptomic studies of MPS type I fibroblasts (Gaffke et al., 2021; Gaffke et al., 2020).
Pathway analysis of phosphoproteins
To obtain insight into the signaling events, the phosphorylated proteins in MPS IH/S were analyzed using the Cytoscape tool (Bindea et al., 2009). The phosphoproteome revealed enrichment of pathways involved in signaling by Rho GTPases, miro GTPases, RHOBTB3, and CDC42 GTPase cycle, SUMOylation of DNA replication proteins, apoptotic cleavage of cellular proteins, signal transduction, axon guidance, and nervous system development (Table 1).
Pathways Found to Be Enriched in Hurler–Scheie Syndrome
Important mediators involved in signaling pathways by Rho-family GTPases such as RANBP2, LMNB1, ANLN, and PLEKHG3 are hyperphosphorylated, and myosin light chain kinase (MYLK), PGRMC2, PCDH7, and DOCK1 are hypophosphorylated. Rho GTPases regulate several cellular processes including cytoskeletal organization, cell morphology, gene transcription, cell cycle progression, and cell adhesion (Ivetic and Ridley, 2004; Lin and Zheng, 2015). With an odds ratio of 3.89671 (p = 7.02E-07), signaling by Rho GTPases underscores its critical involvement in disease-associated biological processes, and the proteins within this pathway are significantly represented in our dataset.
The proteins involved in the apoptotic cleavage of cellular proteins, including ACIN1, LMNB1, and TJP2, are hyperphosphorylated, whereas GSN exhibits a decreased level of phosphorylation. Hyperphosphorylated proteins ACIN1, LMNB1, and TJP2 can potentially impact the breakdown of cellular proteins that are part of the apoptosis process, ultimately leading to disturbances in the apoptotic signaling cascade (Dix et al., 2012; Jana et al., 2021; Tang et al., 2021). (Dix et al., 2012). In general, dysregulation of kinases and proteins in the identified pathways indicates the possibility of disruptions in critical physiological processes. Understanding how these dysregulated proteins interact with their associated pathways could reveal key insights into the molecular mechanisms driving the development of MPS IH/S. This information could potentially lead to the development of therapeutic interventions that are designed to target these specific pathways.
Protein–protein interaction network analysis
Drawing conclusions solely from changes in phosphorylation can be challenging due to the limited functional information regarding whether phosphorylation at a specific location on a given protein has an inhibitory or excitatory effect (Rudolph et al., 2016). Network-based methods are used to analyze phosphorylation data to obtain functional information regarding proteins. This was done by examining the changes in phosphorylation that occur in the network neighborhood (Rudolph et al., 2016). The PPI network was generated using the complete list of dysregulated phosphoproteins including 116 upregulated and 82 downregulated proteins. On generating the PPI network using the STRING database, 146 dysregulated phosphoproteins along with 4 (C17orf85, SDPR, PRKCDBP, and HN1) nonphosphorylated proteins were found to be interacting as depicted in Figure 3A. Among these, 27 were enzymes and are represented as yellow nodes in the network (Fig. 3A). CDK1 and TOP2A were identified as the top 10 hub nodes in the network along with ANLN, MCM2, MCM4, CENPF, MKI67, LMNB1, CEP55, and TPX2.
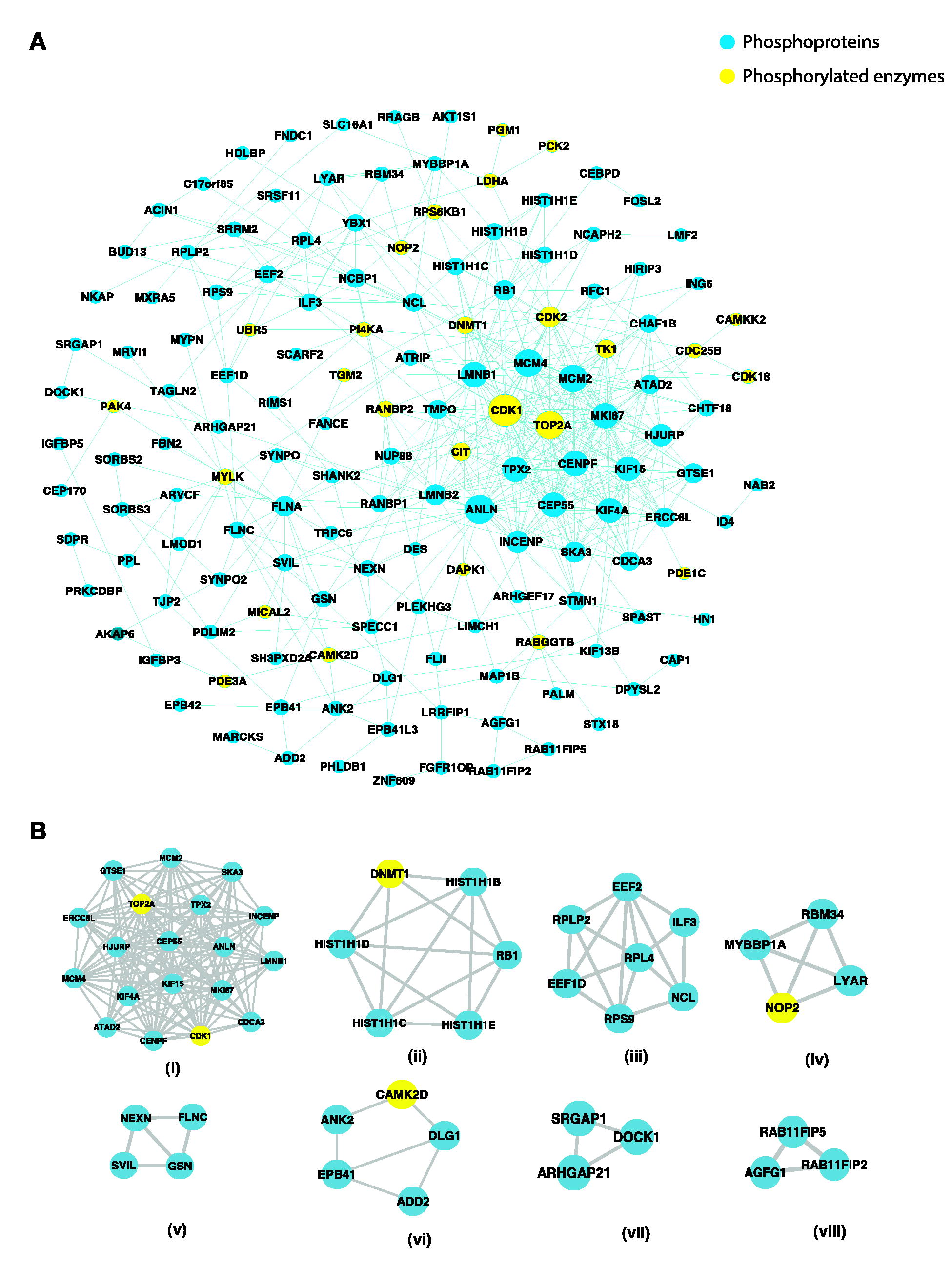
Signaling networks in Hurler–Scheie syndrome (MPS IH/S).
In addition, the PPI network was analyzed using the MCODE algorithm to identify dense networks within the PPI network. Eight clusters were identified, representing highly interacting protein clusters in the PPI network (Fig. 3B). The key pathways involved in these clusters are (i) mitotic spindle assembly and mitotic cytokinesis, (ii) the apoptotic execution phase, (iii) protein synthesis, (iv) nucleolar organization, (v) cytoskeletal organization, (vi) regulation of action potential and spectrin binding, (vii) Rho GTPase signaling, and (viii) intracellular trafficking.
Changes in the cell cycle processes in MPS IH/S
In the subnetwork [Figure3B (i)], the top hub nodes CDK1 and TOP2A are known to interact with KIF15, INCENP, KIF4A, TPX2CEP55, and ANLN. These protein interactions were related to mitotic spindle assembly (GO:0090307) and mitotic cytokinesis (GO:0000281), which highlights the dysregulation of the cell cycle (GO:007049) in MPS IH/S (Supplementary Table S5) (Gaffke et al., 2021). Key M-phase proteins, such as KIF4A (S801), INCENP (S306), CEP55 (S96), and ANLN (S642) are involved in mitotic cytokinesis (Dephoure et al., 2008; Fabbro et al., 2005; Kurasawa et al., 2004). Recent transcriptomic studies indicate the dysregulation of expression of genes coding for proteins involved in cell cycle across all types of MPS fibroblasts (Brokowska et al., 2022). Flow cytometric analysis revealed that the cell cycle processes were disrupted, with increased proportions of cells in the G0/G1 phase observed in the majority of MPS disorders and decreased proportions of cells in the G2/M phase observed across all MPS types (Brokowska et al., 2022).
Hyperphosphorylation of INCENP (S306) can disrupt prometaphase progression and cytokinesis (Mackay et al., 1998). The hyperphosphorylation of key proteins, such as TOP2A, RANBP2, INCENP, and NUP88, suggests significant disruptions in DNA replication, repair, and cell cycle processes (Fernandez-Miranda et al., 2010; Lee et al., 2018; Ma et al., 2021; Moreno-Onate et al., 2020). These disruptions could further contribute to genomic instability, impaired cell division, and possibly increased cellular stress, potentially leading to cell death.
Altered apoptotic processes in MPS IH/S
Apoptosis is achieved by caspase-mediated cleavage of several vital proteins involved in cell adhesion and maintenance of the cytoskeletal structure (Fischer et al., 2003; Wee et al., 2007). The key pathway involved in cluster (ii) (Fig. 3B) was related to the apoptotic execution phase. Hyperphosphorylation of DNMT1 (S143) can affect its activity and interaction with other proteins involved in response to DNA damage (Esteve et al., 2011; Ha et al., 2011).
Hyperphosphorylation of histone H1 variant proteins [Figure 3B (ii)] can affect the transcription of genes involved in DNA damage response (Contreras et al., 2003; Izzo and Schneider, 2016). Furthermore, the induction of apoptosis is associated with the hyperphosphorylation of H1 variant proteins, which undergo rapid dephosphorylation during apoptotic DNA fragmentation (Kratzmeier et al., 2000; Talasz et al., 2002). In addition, hyperphosphorylation of RB1 (T826) is involved in the apoptotic execution phase in mitochondria (Hilgendorf et al., 2013; Sanidas et al., 2019).
Apoptotic cleavage of cellular proteins is a key step in programmed cell death, involving the rearrangement of tight junctions, adherens junctions, and desmosomes (Cirillo et al., 2008; Weiske et al., 2001). ACIN1 confers binding of RNA to the apoptosis- and splicing-associated protein complex involved in splicing of the exon junction complex (Michelle et al., 2012). In addition to splicing, ACIN1 is also involved in chromatin condensation during apoptosis (Schwerk et al., 2003). Hyperphosphorylation of ACIN1 may enhance chromatin condensation and accelerate apoptotic processes.
CDK1 phosphorylates Lamin B1 (LMNB1), a structural component of the nuclear lamina, playing a crucial role in the maintenance of nuclear integrity (Burke and Stewart, 2013; Kuga et al., 2010). The hyperphosphorylation of LMNB1 at serine residues S391 and S393 may facilitate nuclear disassembly and enhance apoptotic processes (Machowska et al., 2015; Torvaldson et al., 2015). In addition to nuclear integrity, the preservation of cellular barriers is essential for tissue homeostasis. Tight junctions between cells play a role in maintaining the integrity of the epithelial and endothelial barriers (Tsukita et al., 1999). Hyperphosphorylation of tight junction protein TJP2, can disrupt these junctions, leading to increased cell permeability and susceptibility to apoptosis (Seth et al., 2007). Collectively, these insights underscore the crucial functions of phosphorylation in modulating nuclear stability and cellular adhesion, hence affecting cell viability and functionality.
Cytoskeletal and structural abnormalities
The cluster (v) cytoskeletal organization [Figure3B (v)] subnetwork suggests a potentially altered structural integrity affecting the stability and motility of the cell. The interaction between nexilin (NEXN), filamin C (FLNC), supervillin (SVIL), and gelsolin (GSN) are closely related to cytoskeletal organization (GO:0007010) with each playing a role in the regulation of actin filament dynamics (GO:0030029, GO:0030036) and structural integrity of the cell (Nag et al., 2009; Oh et al., 2003, Wang et al., 2005). Dysregulation of proteins such as NEXN, FLNC, SVIL, and GSN within this network cluster (v) could impair fibroblast motility, stability and structural organization contributing to progressive tissue damage and fibrosis observed in MPS IH/S patients.
The Rho signaling pathway is a complex intracellular signaling network that regulates various cellular processes, including cytoskeletal dynamics, cell adhesion, cell cycle progression, and cell migration (Jaffe and Hall, 2005; Rodenburg and van Buul, 2021). Rho GTPases regulate actin cytoskeleton organization and influence cell adhesion and migration (Lee and Dominguez, 2010).
Multiple kinases are differentially phosphorylated in MPS I H/S
A total of 239 kinases were identified in the phosphoproteomic analysis of MPS I H/S fibroblast lysates (Fig. 4A; Supplementary Table S6). Twelve kinases were found to be dysregulated in our dataset. Of these, five kinases were hyperphosphorylated (CDK1, CDK2, CIT, PAK4, and WNK4), and seven kinases were hypophosphorylated (CAMK2D, CAMKK2, CDK18, DAPK1, MYLK, RPS6KB1, and SPEG).
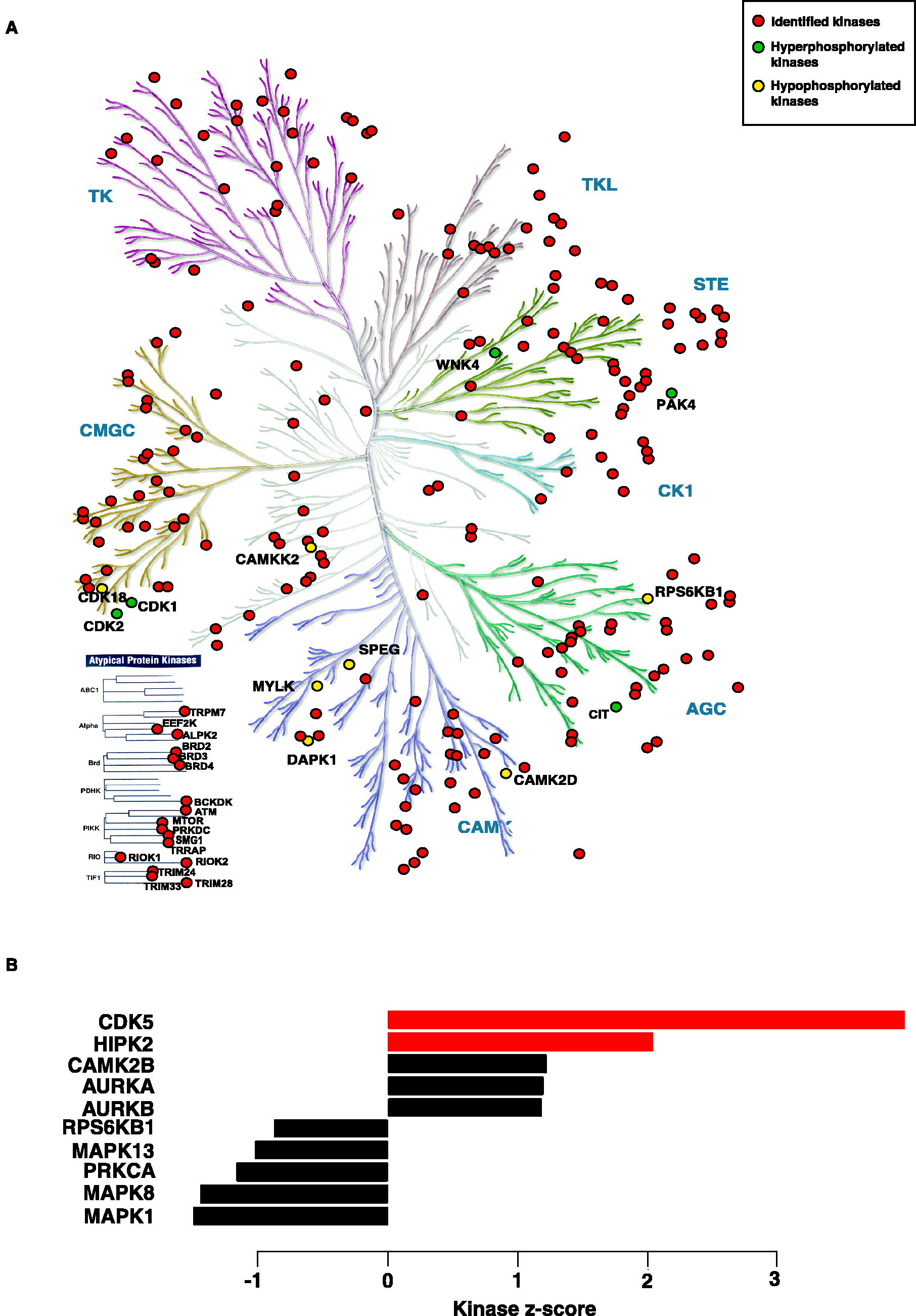
Kinases in Hurler–Scheie syndrome (MPS IH/S).
A key regulator of the cell cycle, CDK1, is essential for cell cycle progression, DNA replication, and DNA damage repair (Liao et al., 2017). Dysregulation of CDK2, which is involved in promoting nascent DNA synthesis, can affect DNA replication processes (Li et al., 2012). One of the hypophosphorylated kinases, CAMKK2, acts as a signaling hub that phosphorylates downstream targets such as AMPK and CAMK types I and IV. Dysregulation of CAMKK2 affects various cellular processes, including cell proliferation and apoptosis (Profeta et al., 2019).
DAPK1, an important regulator of cell death and autophagy, is a stress-responsive serine/threonine kinase that is dysregulated and affects apoptosis (Singh et al., 2016). Hypophosphorylated kinases such as MYLK, SPEG, and RPS6KB1 are involved in smooth muscle contraction, muscle development, and protein synthesis, respectively (Basu and Proweller, 2016; Luo et al., 2021; Ruvinsky and Meyuhas, 2006). Overall, specific dysregulated kinases (such as CDK1, CDK2, CAMKK2, DAPK1, MYLK) identified by phosphoproteomic analysis of MPS I H/S fibroblast lysates are implicated in disruption of cellular processes such as cell cycle regulation, DNA replication/repair and apoptosis, as well as muscular function.
Kinase substrate enrichment analysis in MPS I H/S
Kinase substrate enrichment analysis, a method for the systematic profiling of kinase pathway activities, was performed to predict the upstream kinases responsible for the observed differential phosphorylation (Wiredja et al., 2017). Two kinases, CDK5 (z-score = 3.98 and p = 3.43 × 10−1) and HIPK2 (z-score = 2.04, p < 0.05), were predicted to be activated (Fig. 4B) in MPS IH/S. Kinase-substrate links are listed in Supplementary Table S6c. CDK5 and HIPK2 were responsible for the phosphorylation of 2 (MAP1B, DPYL2) and 1 (MCM4) downstream proteins, respectively.
CDK5, a proline-directed serine/threonine kinase participates in a range of cellular processes, such as cell cycle regulation, apoptosis, and cytoskeletal dynamics, which are relevant to fibroblasts (Contreras-Vallejos et al., 2012). Both MAP1B and DPYL2 are involved in microtubule stabilization, actin filament binding, cytoskeletal rearrangements, and cellular migration, which are critical processes in fibroblast function under stress or pathological conditions (Kuter et al., 2016; Togel et al., 1998).
Homeodomain-interacting protein kinase 2 (HIPK2) is a DNA damage-responsive serine/threonine kinase that activates various cellular processes, including apoptosis (Calzado et al., 2007; Kuwano et al., 2016). HIPK2’s role in DNA damage response and cell cycle regulation may affect MCM4 phosphorylation directly or indirectly through its downstream effects on Cdc7 or CDK2 activity. During DNA replication, MCM4 is phosphorylated by Cdc7 kinase, allowing it to bind to Cdc45 on the chromatin (Masai et al., 2006). Furthermore, CDK2 inhibition affected MCM4 phosphorylation at CDK sites during the cell cycle (Komamura-Kohno et al., 2006). Further studies are required to elucidate the mechanisms and functional consequences of HIPK2-mediated phosphorylation of MCM4.
Conclusions
The present comprehensive analysis of the proteome and phosphoproteome in MPS IH/S offers new and valuable insights into the molecular underpinnings of the disease, revealing that widespread GAG accumulation can lead to secondary alterations in protein expression and phosphorylation, reflecting broader pathophysiological changes. The identification of proteins such as CDK5, MAP1B, and DPYL2 in fibroblasts from patients with MPS IH/S suggests that these molecules play important roles in cellular processes such as cytoskeletal organization, cell cycle regulation, and intracellular signaling. Their altered expression could contribute to the broader cellular dysfunction observed in MPS IH/S, potentially affecting the cell structure, division, and communication pathways.
Moreover, these findings may serve as indirect indicators of neuronal dysfunction and provide insight into the multisystemic effects of MPS IH/S. Additional research is necessary to elucidate the functional characterization of neuronal cells, which may provide further insights into the underlying neurological disturbances. Further translational clinical omics studies are warranted to facilitate the development of innovative diagnostics and therapeutics for patients with MPS IH/S.
Footnotes
Acknowledgments
The authors thank Mayank Saraswat for his input during initial planning of the study.
Authors’ Contributions
M.G.R. and A.P. conceptualized and designed the study. M.G.R., J.S., K.T.S.P., and K.B.G. curated the data. Formal analysis was carried out by M.G.R., K.T.S.P., K.B.G., and J.S. Funding acquisition was done by A.P. Investigation was carried out by M.G.R., K.G., N.J., and J.S. Methodology was taken care by M.G.R., K.G., N.J., and A.P. Project administration was carried out by A.P. Resources were taken care by M.G.R. and A.P. Software analysis was done by M.G.R., K.T.S.P., K.B.G., and J.S. Writing (original draft, reviewed, and edited) was done by M.G.R. Critical review of the article done by J.S., R.K., and A.P. All authors have read and approved the final version of the article.
Author Disclosure Statement
The authors declare they have no conflict of interest.
Funding Information
This study was supported by a grant from DBT/Wellcome Trust India Alliance entitled “Center for Rare Disease Diagnosis, Research, and Training” (
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.