
Abstract
Climate emergency and ecological sustainability call for new ways of thinking livestock health, including the dairy cattle. This study unpacks the genetic diversity and selection sweeps of Sahiwal cattle in relation to adaptability, production, and disease resistance. Using nucleotide diversity (π) calculated from 10 kb windows across the genome with VCFtools, 716 regions of genetic diversity were identified across 29 chromosomes, and importantly, with chromosome 15 showing the highest density. A total of 92 quantitative trait loci (QTL) linked genes were analyzed, with chromosome 1 harboring the highest number. Trait association analysis using the Cattle QTL database showed that 14 genes were linked to production traits, 10 to reproduction traits, and 8 to disease susceptibility. Notable genes included CSMD2 and EFNA1, which influence milk production traits such as fat percentage and yield, and PCBP3 and SGCD, which affect reproductive traits. Additionally, the genes TBXAS1 and ASTN2 were associated with disease traits such as bovine respiratory disease and sole ulcers. Selection sweeps, identified using Tajima’s D, revealed 728 sweeps across the genome, with chromosomes 6 and 8 showing the highest frequencies. These sweeps indicate regions under strong selective pressure, likely due to the breed’s adaptation to arid environments and specific trait selection. The present study highlights how genetic diversity and selection sweeps contribute to Sahiwal cattle’s adaptability, production efficiency, and disease resistance. The insights reported here provide a foundation for livestock health and targeted breeding strategies in the case of Sahiwal cattle under diverse ecological conditions such as tropical climate.
Introduction
The intensifying climate crisis presents significant challenges to global livestock health, productivity, and welfare, highlighting the urgent need for innovative and sustainable strategies in animal agriculture. Dairy cattle, as a key component of global food systems, are particularly vulnerable to the adverse effects of climate change, including rising temperatures, water scarcity, heat stress, and increased disease pressures. In this context, there is growing interest in harnessing the genetic potential of resilient cattle breeds to enhance climate adaptability and ensure the sustainability of dairy production worldwide (Kaplan et al., 1989; Sun et al., 2024).
Recent advancements in livestock genomics have identified key genetic markers in various dairy cattle breeds associated with enhanced immune responses, metabolic efficiency, and production traits (Pushpa et al., 2023a). For instance, genomic studies on indigenous breeds have revealed the presence of adaptive genes linked to heat tolerance, disease resistance, and improved fertility under stressful environmental conditions. These insights have far-reaching implications not only for improving local breeding programs but also for addressing broader global challenges in planetary veterinary medicine. Understanding the genetic basis of such adaptive traits enables the development of targeted breeding strategies to enhance the resilience and productivity of dairy cattle in diverse agro-climatic regions (Brito et al., 2021; Marchioretto et al., 2023; Pushpa et al., 2023b, 2024).
Integrating genomic insights from resilient dairy cattle breeds into global livestock management frameworks offers a strategic pathway to mitigate the impacts of climate change on animal agriculture. This approach aligns with emerging paradigms in planetary health, which emphasize the interconnectedness of human, animal, and environmental well-being. Strengthening the genetic resilience of dairy cattle through advanced breeding programs and genomic research not only enhances food security and production efficiency but also supports ecological sustainability in the face of a rapidly changing global climate.
Domestication and selective breeding have profoundly influenced the genomes of livestock, including cattle. Artificial selection, as opposed to natural selection, can induce rapid changes in the genome, particularly in regions associated with economically and agriculturally significant traits. These changes often manifest as selection sweeps, where specific regions of the genome exhibit reduced genetic variability due to the fixation of advantageous alleles. This process, known as the hitchhiking effect, not only impacts the selected traits but also affects neutral alleles at linked loci (Kaplan et al., 1989; Smith and Haigh, 1974). Identifying these selection signatures is essential for understanding the genetic basis of traits that are crucial for the productivity, resilience, and adaptability of cattle (Kohn et al., 2000; Nielsen et al., 2005).
Sahiwal is a well-known breed belonging to the humped zebu cattle group (Bos indicus), originates from the northwestern region of the Indian subcontinent (Magotra et al., 2019; Muhuyi et al., 1999; Yadav et al., 2023). In addition to their high milk production, Sahiwal cattle are uniquely adapted to hot and humid climates and exhibit resistance to tropical diseases, ticks, and parasites (Singh et al., 2005). Their milk is notable for its high fat content (4.6–5.2%) and solid non-fat (8.9–9.3%) (Joshi et al., 2001). Due to these attributes, Sahiwal cattle are utilized in cross-breeding programs globally to enhance milk production and environmental endurance (Illa et al., 2021).
However, the pure indigenous germplasm of Sahiwal is dwindling due to indiscriminate and unplanned cross-breeding, intensified production systems, and purpose-driven farming practices (Magotra et al., 2020; Magotra et al., 2015; Srivastava et al., 2019; Yadav et al., 2020). The extensive human-mediated selection in Sahiwal cattle has likely led to substantial genomic alterations, including the creation of selective sweeps where beneficial mutations have been driven to fixation, reducing genetic diversity in adjacent genomic regions (Berry et al., 2017; Kristensen et al., 2015; Ramey et al., 2013). These selective sweeps provide valuable insights into the historical and ongoing selection pressures acting on the breed, shedding light on the genetic factors contributing to their desirable traits.
Recent advances in molecular genetic techniques, particularly the development of next-generation molecular markers, have greatly enhanced the scope of bovine genome research. Among these, reduced representation sequencing approaches, commonly known as restriction site associated DNA sequencing (RADseq), stand out as cost-effective and efficient methods for targeting specific genome subsets using restriction enzymes, thereby allowing for greater coverage depth per locus (Dennis et al., 2003; Luikart et al., 2003).
RADseq loci, typically conserved within populations and found in both coding and non-coding regions, have been utilized in various genetic analyses in cattle, including the estimation of genetic diversity (Hosoya et al., 2018), the analysis of genetic divergence among breeds (Malik et al., 2018), genome-wide association studies (Elshire et al., 2011), the design of breed-specific SNP panels (Lopez de Heredia et al., 2020) and the identification of selection signatures (Kour et al., 2022).
While RADseq techniques have been extensively applied to taurine cattle breeds, studies focusing on indigenous breeds like Sahiwal are scarce. Selection signatures, which are specific DNA-level variations resulting from changes in both selected and neutral loci under selection, are critical for understanding the genetic mechanisms underlying important traits (Kreitman, 2000; Magotra et al., 2016; Magotra et al., 2015). Given the unique adaptations of Sahiwal cattle to tropical environments and their valuable milk attributes, it is likely that their genomic regions have been subject to intense selective pressure for centuries. However, research in this area remains limited.
The aim of the present study was to address this knowledge gap by conducting a genome-wide scan to identify selection signatures in Sahiwal cattle using the ddRAD sequencing approach. By exploring these candidate genes, the research sought to contribute to the conservation and improvement of this vital indigenous breed.
Materials and Methods
The current research was conducted on a sample size comprising eight Sahiwal cows. For the Sahiwal cows, blood samples were collected randomly from animals housed at the Cattle Breeding Farm, Lala Lajpat Rai University of Veterinary & Animal Sciences, located in Hisar, Haryana, India. The blood samples of the animals were collected in a sterile vacutainer tube coated with 0.5% ethylene diamine tetraacetic acid (EDTA). The study and blood collection were conducted with the relevant guidelines and regulations as approved by the Institutional Animal Ethics Committee (IAEC, Hisar).
The genomic DNA was isolated from blood samples by the phenol:chloroform:isoamyl alcohol extraction method with slight modifications.
Library preparation
Genomic DNA (1 µg) meeting the quality check parameters of more than 100 ng/µL concentration and an OD260/OD280 ratio of 1.7–1.9 was subjected to library preparation following the standard ddRAD protocols described by Peterson et al. (2012). The genomic DNA was double-digested using restriction enzymes Sph I and Mlu I, followed by cleanup using Ampure beads. Suitable adapters were ligated using T4 DNA ligase, and the ligated products were pooled and cleaned up. Size selection was performed using 2% agarose gel electrophoresis, and the size-selected products were enriched through PCR amplification, which also incorporated Illumina-specific adapters and flow cell annealing sequences. The quality of the libraries was assessed using a bioanalyzer, and the libraries were subsequently pooled for next-generation sequencing (NGS) on an Illumina sequencing platform. The ddRAD library preparation and sequencing were conducted by Redcliffe labs private limited, Noida (UP), India.
Bioinformatics analyses
Read processing
The raw sequencing reads generated from the Sahiwal cattle samples were first demultiplexed to assign reads to their respective samples. Custom perl scripts were utilized for this demultiplexing process, allowing for the efficient isolation of sample-specific reads based on their unique barcodes. The scripts permitted up to one mismatch in the barcode sequence, providing flexibility while maintaining the accuracy of sample identification. This demultiplexing step ensured that each read was appropriately associated with its originating sample, which is crucial for downstream genomic analyses.
After demultiplexing, the reads underwent a thorough preprocessing step to remove low-quality bases and sequences that might affect the accuracy of subsequent analysis. Prinseq-lite version 0.20.4 (Schmieder and Edwards, 2011) was employed for this purpose. Prinseq-lite was used to trim low-quality bases at both the 5′ and 3′ ends of the reads, as well as to remove regions exhibiting base biases, which are common near the beginnings or ends of sequencing reads. This process minimized the inclusion of sequencing errors and improved the overall quality of the dataset.
Additionally, the Illumina adapter sequences from the 5′ and 3′ ends were trimmed to prevent non-genomic sequences from being included in the final dataset. Adapter contamination is a common issue in NGSdata, and its removal ensures that only biologically relevant sequences are retained for further analysis.
To confirm the quality of the processed reads, the FastQC tool was employed. FastQC provided a comprehensive report on the read quality, highlighting key metrics such as per base sequence quality, GC content, and sequence duplication levels. This quality control step was vital in ensuring that the data met the necessary standards for high-confidence downstream analyses.
Subsequently, the processed reads were used to identify restriction site associated DNA (RAD) loci using the process radtags module from the stacks software package (Catchen et al., 2013). Process radtags was specifically designed for analyzing RADseq data, a technique commonly used in population genomics. This step enabled the identification and extraction of RAD loci from the sequence reads, filtering out any low-quality reads and demultiplexing errors. Stacks helped to ensure that only high-quality RAD loci were retained for alignment and subsequent genetic analysis.
Alignment
Following the quality filtering process, the filtered reads were subjected to alignment using the Burrows-wheeler alignment tool within the dDocent framework. This alignment step involved aligning the filtered reads against the reference genome, which in this study is the Bos taurus (cattle) reference genome. The specific reference genome utilized in this study can be accessed via the provided URL: https://www.ncbi.nlm.nih.gov/assembly/GCF_002263795.1/. The alignment step was crucial for identifying and mapping genetic variants, which were subsequently used to generate a Variant Call Format (VCF) file. The VCF file was created using FreeBayes, a Bayesian genetic variant detector designed to handle high-throughput sequencing data. The VCF file, produced at RD10, facilitated the detection of selection signatures by providing detailed information on genetic variants, thereby aiding in the elucidation of genomic differences in Sahiwal cattle.
Nucleotide diversity (π) and Tajima’s D
Nucleotide diversity (π) and Tajima’s D were calculated to evaluate genetic variation and selection pressures in Sahiwal cattle using vcftools version 0.1.16 (Danecek et al., 2011). Nucleotide diversity (π) measures the average pair-wise differences between sequences within a population, providing an estimate of genetic variation. Higher nucleotide diversity indicates greater genetic variation, while lower values suggest reduced variation due to selective sweeps or population bottlenecks. Tajima’s D assesses the neutrality of mutations by comparing the number of segregating sites to the average number of pair-wise differences; positive values of Tajima’s D indicate balancing selection or population contraction, whereas negative values suggest purifying selection or population expansion. The analysis was conducted separately for each chromosome to provide detailed insights into genomic patterns.
Genotypic data processing
The VCF file was processed to ensure high data quality and reliability. Variants with a minor allele frequency below 0.05 were excluded to focus on more prevalent variants. Variants with more than 50% missing data were removed to maintain data integrity. Variants showing deviations from Hardy-Weinberg equilibrium at p < 0.0001 were also excluded to minimize the influence of potential genotyping errors. Variants were required to have a minimum quality score of 20 to ensure high confidence in the calls. Genetic diversity was computed as the average number of nucleotide differences per site between pairs of sequences, providing an estimate of genetic variation within the population. Tajima’s D was calculated to identify deviations from neutrality by comparing the number of segregating sites with the average number of nucleotide differences. This statistic aids in detecting signals of selection, population expansion, or demographic changes.
A sliding window analysis was employed to examine the distribution of genetic diversity and Tajima’s D along the genome. Windows of 50,000 base pairs were used, with a step size of 20,000 base pairs. This approach allowed for a detailed assessment of genetic diversity and selection sweeps across different genomic regions, revealing variations and trends in the genetic metrics.
Identification of functional genes and quantitative trait loci
The genomic regions were considered as significant if q value is lower than 0.05 for adjacent SNPs. The boundaries of the genomic regions were determined from the SNP with a q value greater than 0.1. A q value threshold of <0.05 was chosen to control the false discovery rate (FDR) and reduce the likelihood of type I errors. The gene and quantitative trait loci (QTL) annotations were performed using R package Genomic Annotation in Livestock for positional candidate Loci (GALLO) (Fonseca et al., 2020). The gene and QTL annotation files (.gtf and .gfffiles) derived from the ARS-UCD1.2 assembly and Animal QTL Database (Hu et al., 2013) were used for the gene and QTL identification, respectively. The QTL enrichment analysis was also performed for all the QTLs annotated by the chromosome-based method using the same GALLO package.
A bootstrap method was implemented to correlate the observed and expected number of QTLs per trait from the cattle QTL database with 1000 iterations of random sampling. The calculated p values in the enrichment analysis were also adjusted using FDR (<5%) for multiple testing.
Classification of traits
The Supplementary Table S1 categorizes various traits linked to performance and disease susceptibility in livestock, as detailed in the ANIMALQTL database (Animal QTL Database [animalgenome.org]). The traits are grouped into four main categories: production traits, reproduction traits, disease-associated traits, and miscellaneous traits (covering adaptability, conformation, and milk quality).
Gene ontology
The genes identified were submitted to Database for Annotation, Visualization and Integrated Discovery (DAVID) (DAVID Functional Annotation Bioinformatics Microarray Analysis [ncifcrf.gov]) for gene enrichment analysis (Dennis et al., 2003). The clustering was done based on gene ontology (GO), biological process (BO) terms, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Ashburner et al., 2000; Ogata et al., 1999; Ravinath et al., 2024).
Results
Genome-wide identification of genetic diversity regions in Sahiwal cattle
In present study, genetic diversity (π) in Sahiwal cattle was assessed using vcftools by analyzing 10 kb windows across the genome. Supplementary Table S2 summarizes the findings on chromosome-wise genetic diversity. A total of 716 genetic diversity regions were identified across 29 chromosomes with chromosome 15 showing the highest diversity regions (51). Among the 92 QTL-associated genes at chromosome 1 had the most (7) highlighting the complex genetic landscape of Sahiwal cattle.
In this investigation of genetic diversity regions within Sahiwal cattle, the association of genes with targeted traits was explored using the cattle QTL database. Table 1 presents the distribution of genes across various trait categories within these regions. Among the 92 genes identified, 14 were associated with production traits, 10 with reproduction traits, and 8 with disease-associated traits. Notably, 31 genes were linked to miscellaneous traits reflecting the diverse genetic landscape of Sahiwal cattle. Additionally, 10 genes were associated with multiple traits, including production, reproduction, disease, and conformation traits, underscoring the intricate interplay of genetic factors influencing multifaceted phenotypic characteristics in this breed.
Number of Genes Associated with Targeted Traits Identified in Genetic Diversity Regions in Sahiwal Cattle
The genetic diversity regions in Sahiwal cattle harbor genes crucially associated with various production, reproduction, and disease traits. In Table 2, genes such as CSMD2, EFNA1, and CRACR2A were identified influencing milk fat percentage, milk protein percentage, and milk fat yield, respectively, highlighting their roles in regulating milk production traits. Moreover, Table 2 unveils genes like PCBP3, SGCD, and WWOX linked to traits such as interval to first estrus after calving, first service conception and pregnancy rate, shedding light on their significance in reproductive performance. Similarly, Table 2 elucidates genes such as TBXAS1, ASTN2, and NF1 associated with traits like bovine respiratory disease susceptibility, sole ulcer, and bovine tuberculosis susceptibility providing insights into disease susceptibility mechanisms in Sahiwal cattle. These findings emphasize the intricate genetic architecture governing diverse phenotypic traits in this cattle breed.
Genes Associated with Production, Reproduction, and Disease Traits Identified in Genetic Diversity Regions in Sahiwal Cattle
Genome-wide identification of selection sweeps in Sahiwal cattle
In the genomic analysis of Sahiwal cattle, Tajima’s D was employed to identify selection sweeps, providing insights into the evolutionary dynamics of the population. Notably, a total of 728 selection sweeps were detected across the genome, with varying frequencies observed on different chromosomes. Chromosomes 6 and 8 exhibited the highest number of selection sweeps, with 38 and 40 sweeps, respectively. Conversely, chromosomes 12 and 20 displayed the lowest frequencies, with only 5 and 8 sweeps, respectively.
In the examination of putative selective sweep regions within Sahiwal cattle, Supplementary Table S3 delineates the chromosome-wise distribution of these sweeps, revealing a total of 728 identified sweeps, with varying frequencies across chromosomes. Furthermore, candidate genes associated with performance and disease traits within these selective sweep regions were explored, as depicted in Tables 3 and 4. Notably, a diverse array of genes linked to production traits, reproduction traits, and disease susceptibility were identified, shedding light on the genetic underpinnings of key phenotypic characteristics in Sahiwal cattle. Specifically, genes such as HERC6, CTNNA2, and PREX1 were found to influence milk fat percentage, milk fat yield, and milk protein yield, respectively, implicating their roles in regulating milk production traits. Additionally, genes like NTNG1, AFAP1, and SGCD were associated with inseminations per conception and first service conception, highlighting their significance in reproductive performance. Moreover, genes such as KCNJ3, DGKI, and CAV1 were linked to bovine respiratory disease susceptibility and other disease traits, providing insights into disease resilience mechanisms in Sahiwal cattle.
Candidate Genes Associated with Performance and Disease Traits Identified in Putative Selective Sweep Regions in Sahiwal Cattle
Candidate Genes for Production, Reproduction, and Disease Traits Identified in Putative Selective Sweep Regions in Sahiwal Cattle
Functional annotation
GO and pathway analysis of genes identified in genetic diversity and selection sweep regions in Sahiwal cattle
The GO enrichment analysis and pathway analysis performed on Sahiwal cattle provide valuable insights into the biological processes, cellular components, molecular functions, and pathways associated with genes found within both genome-wide genetic diversity regions and selection sweep regions (Supplementary Tables S4, S5, S6, S7, S8, S9, S10 and S11). These analyses shed light on the genetic mechanisms underlying various physiological traits and biological functions in Sahiwal cattle (Figs. 1–4).
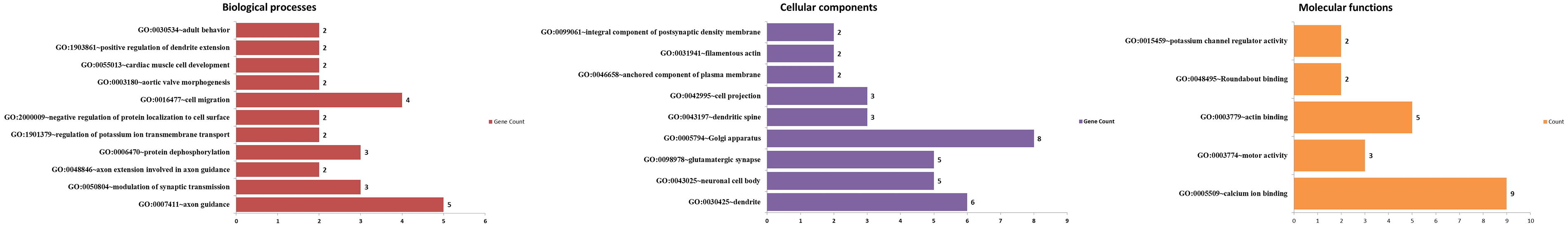
Gene ontology (GO) enrichment analysis of genes identified in regions showing genetic diversity in Sahiwal cattle. Biological processes (in red); cellular component (in purple); molecular function (in yellow). The x-axis lists the GO terms, and the y-axis indicates the number of genes associated with each term.
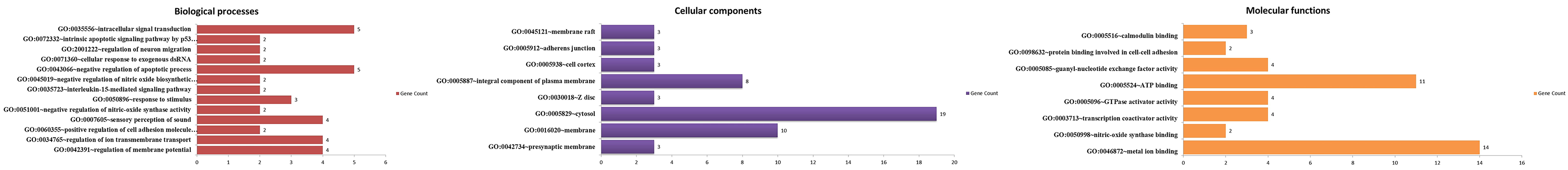
Gene ontology (GO) enrichment analysis of genes identified in selection sweep region in Sahiwal cattle. Biological processes (in red); cellular component (in purple); molecular function (in yellow). The x-axis lists the GO terms, and the y-axis indicates the number of genes associated with each term.
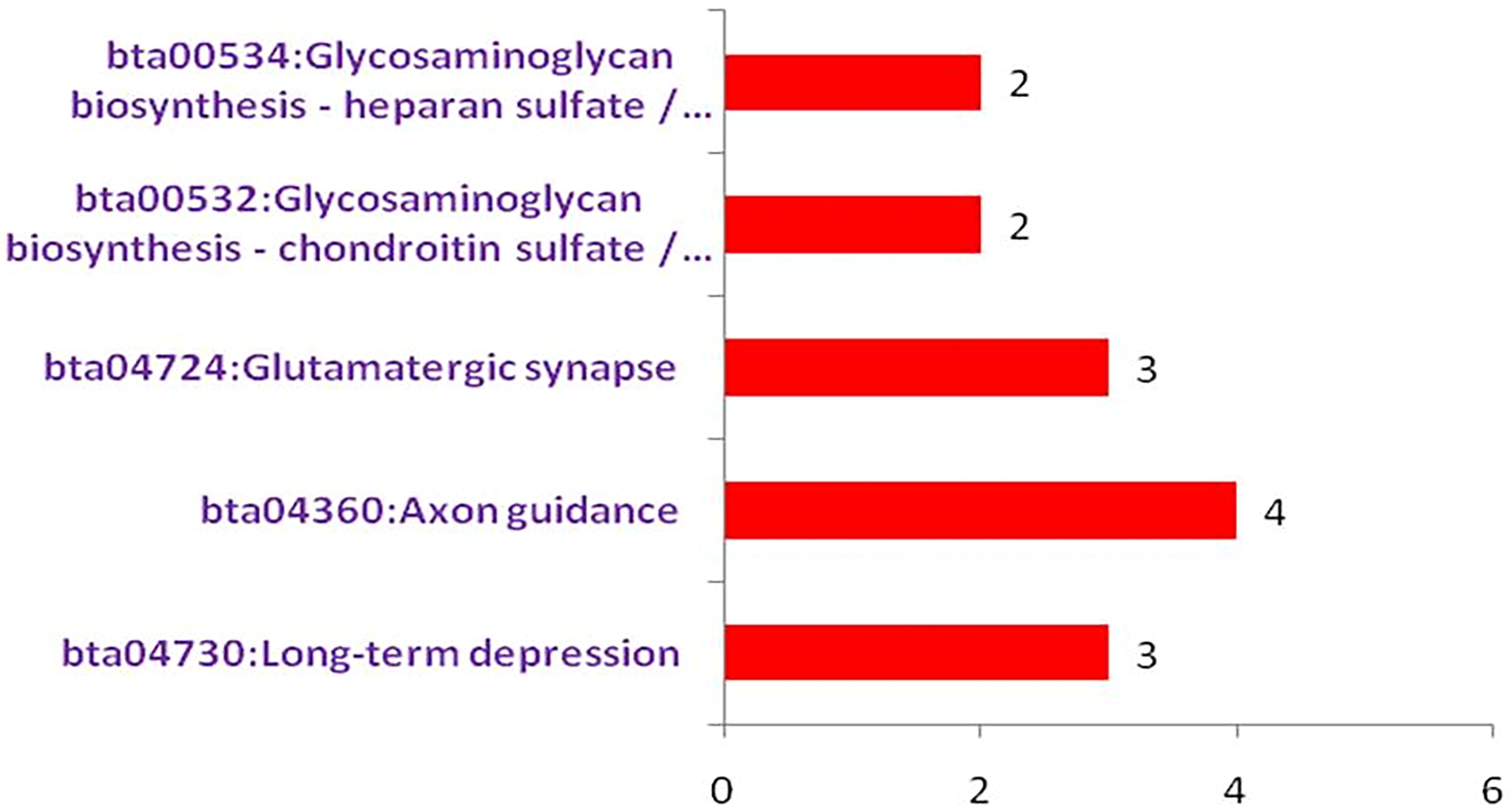
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of genes identified in genetic diversity of Sahiwal cattle. The x-axis shows the number of genes involved in each pathway, and the y-axis lists the enriched pathways.

Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of genes identified in genome-wide selection sweep regions in Sahiwal cattle. The x-axis shows the number of genes involved in each pathway, and the y-axis lists the enriched pathways.
Discussion
Genome-wide genetic diversity analyses in Sahiwal cattle
The genetic diversity of Sahiwal cattle was assessed chromosome-wise, revealing varying degrees of genetic diversity across different chromosomes. A total of 716 genetic diversity regions were identified, spanning all chromosomes except for the X chromosome. The number of genetic diversity regions per chromosome ranged from 7 to 51, with chromosome 15 exhibiting the highest number of genetic diversity regions (51) and chromosome 17 displaying the lowest (7). These regions collectively harbored 92 QTL genes, with chromosome 11 having the highest number of QTL genes (9) and chromosome 27 having no annotated QTL genes. Notably, chromosome number 6 displayed relatively lower genetic diversity with 14 genetic diversity regions only (Supplementary Table S2).
The Manhattan plots (Fig. 5) illustrate the distribution of π values and variant counts across different chromosomes, providing insights into the genetic diversity and variability within the Sahiwal genome. Figure 5a depicts genome-wide genetic diversity (π) along the y-axis, representing the genetic diversity per window, while Figure 5b shows the number of variants per window on the y-axis. The x-axis in both plots represents genomic positions (chromosome-wise). The left plot illustrates the distribution of nucleotide diversity (π) across different genomic regions.
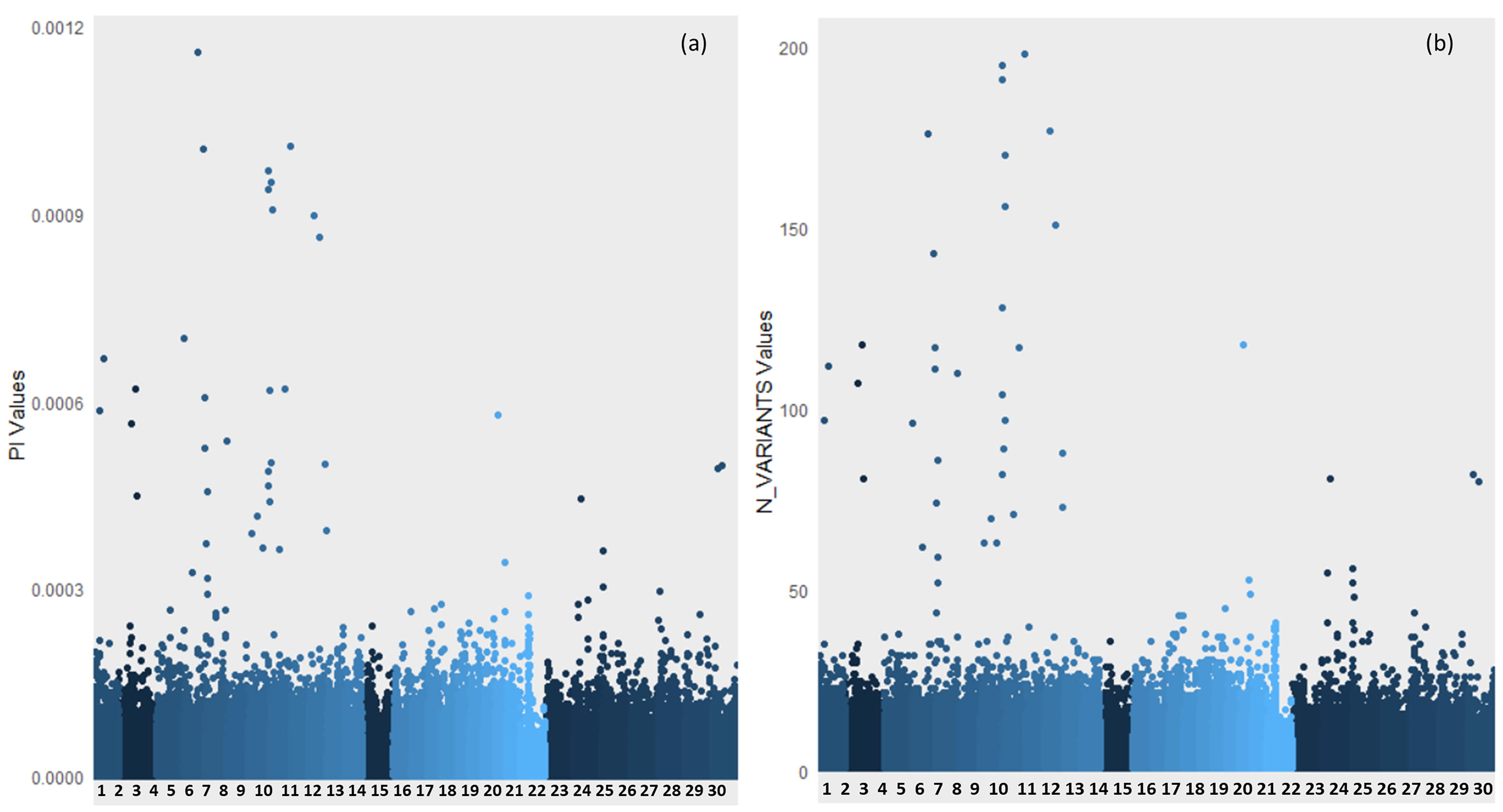
Manhattan plots depicting genome-wide genetic diversity (π) and variant density in Sahiwal cattle. The x-axis represents chromosome numbers. In panel
Nucleotide diversity (π) serves as an indicator of genetic variation within populations. Elevated π values denote higher genetic variation, which may reflect regions under balancing selection or relaxed selective pressure. Conversely, reduced π values suggest regions under purifying selection or selective sweeps, where deleterious alleles are being removed from the population, leading to reduced genetic diversity. This pattern highlights the dynamic nature of selective forces acting on different genomic regions. The right plot depicts the number of genetic variants (N_VARIANTS) identified across genomic windows. Peaks in the distribution of N_VARIANTS values indicate regions with a high density of genetic variants, which could signify hotspots of mutation or recombination. Such regions may represent loci under positive selection or areas with elevated mutation rates. The variation in both π and N_VARIANTS values across chromosomes underscores the heterogeneous nature of the genomic landscape, suggesting that distinct evolutionary pressures are acting on different genomic regions. This comprehensive analysis provides insights into the genomic distribution of genetic diversity in Sahiwal cattle, highlighting potential regions of interest for further investigation into breed-specific traits and adaptation mechanisms.
Genome-wide identification of selection sweeps and associated genes
The genome-wide identification of selection sweeps in Sahiwal cattle, analyzed chromosome-wise, revealed notable patterns of selection across different chromosomes. Figure 6 presents two Manhattan-style scatterplots representing genome-wide variation metrics across different chromosomes. The x-axis in both plots shows genomic positions (Chromosome no.). The left plot (Fig. 6a) displays N_SNPs values on the y-axis, representing the number of SNPs detected in each genomic window. Higher peaks indicate regions with elevated SNP density, which may suggest hotspots of genetic variation or regions under selective pressure. The right plot (Fig. 6b) illustrates Tajima’s D values on the y-axis, which reflect the balance between low- and high-frequency polymorphisms. Positive Tajima’s D values may indicate balancing selection or population contraction, while negative values suggest recent positive selection or population expansion.
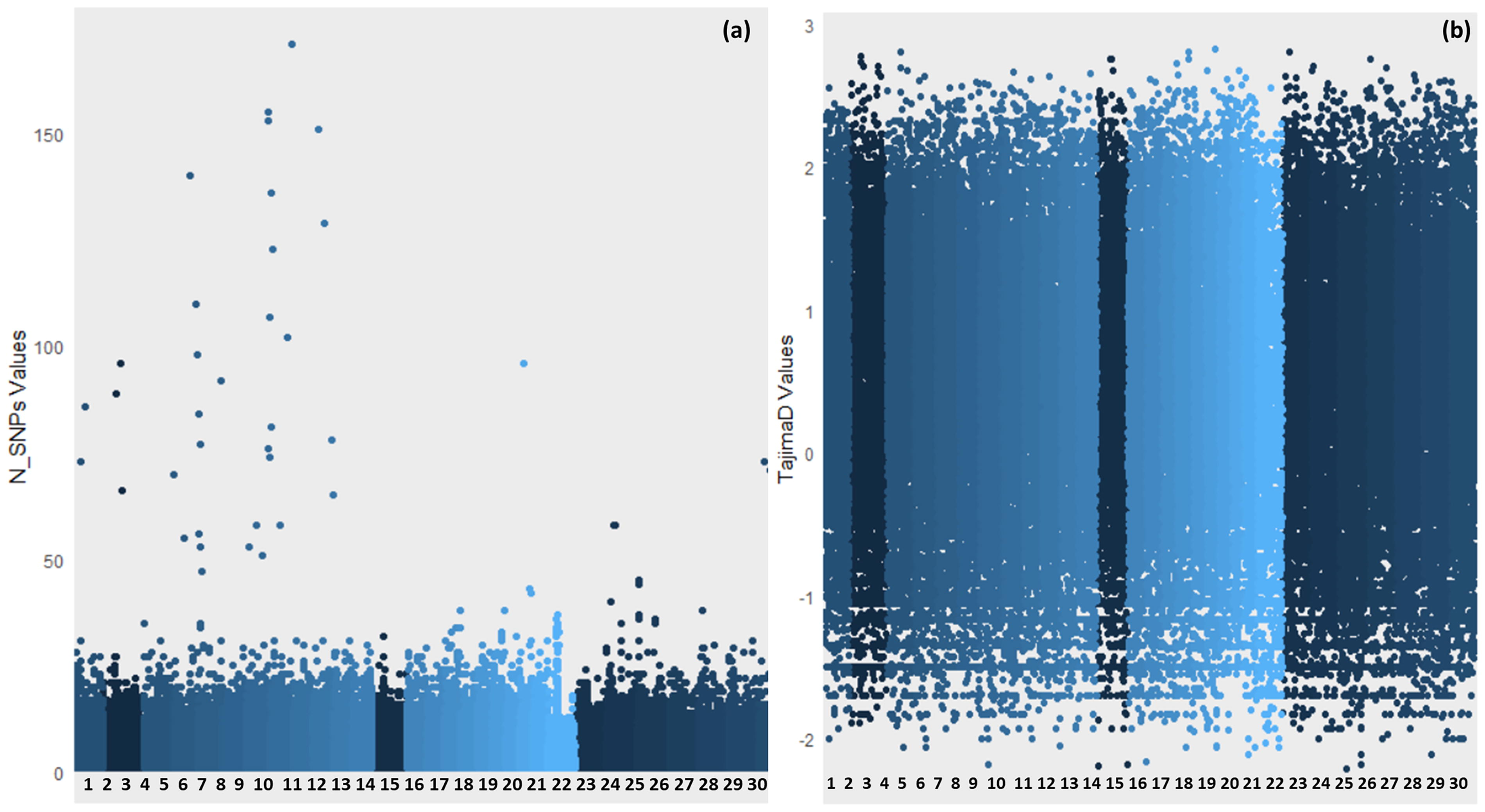
Manhattan plots depicting genome-wide diversity analysis and Tajima’s D values in Sahiwal cattle. The x-axis represents chromosomal and genomic positions. In panel
Earlier studies also analyzed sweep regions utilizing various statistical tools in 12 cattle breeds using bovine SNP50K bead-chip (Rajawat et al., 2022). Tajima’s D test was used to find sequences that show divergence from the neutral model and are based on the site frequency spectrum (1). Supplementary Table S3 presents a summary of these selection sweeps, indicating the number of sweeps identified on each chromosome along with the corresponding number of genes associated with these sweeps. Chromosome 6 exhibited the highest number of selection sweeps with 38, followed closely by chromosome 8 with 40 sweeps. Chromosomes 10, 13, 15, and 17 also displayed relatively high numbers of selection sweeps, ranging from 26 to 39. On the other hand, chromosome X revealed no selection sweep region. In terms of the number of QTL-associated genes associated with these selection sweeps, chromosome 14 stood out with the highest count of eight genes, followed by chromosomes 2, 13, 16, and 17, each with six or seven genes. Chromosomes 21, 27, 28, and 29 exhibited no QTL-associated genes associated with selection sweeps.
In targeted resource population, chromosome 6 emerged as the hotspot for selection sweeps, followed closely by chromosome 8, suggesting significant selection pressure possibly related to production, reproduction, or disease resistance traits. The identification of selection sweeps highlights genomic regions crucial for adaptation to diverse environments and management practices, aiding in the development of breeding strategies for enhancing desirable traits while maintaining genetic diversity.
Candidate genes associated with traits identified in putative selective sweep regions in Sahiwal cattle
Table 3 presents a total of 89 identified candidate genes, categorized according to various trait categories based on the Animal QTL Database (animalgenome.org). The identified genes were assigned to specific traits as per the classifications provided in the database. Notably, Sahiwal cattle exhibit a diverse genetic profile, with genes associated with production traits, reproduction traits, disease susceptibility, and miscellaneous traits. Production traits, such as milk yield and milk fat percentage, are represented by nine genes, indicating genetic factors influencing milk production in this breed. Similarly, reproduction traits are represented by eight genes, suggesting genetic determinants affecting reproductive performance metrics like interval to first estrus after calving and pregnancy rate. Moreover, the analysis identifies 13 genes associated with disease traits, underscoring the importance of genetic factors in determining susceptibility or resistance to various diseases in Sahiwal cattle. Interestingly, some genes are associated with combinations of traits, such as production + reproduction (4 genes) and production + disease (10 genes), indicating potential genetic interactions or pleiotropic effects.
The identification of selection sweep regions in Sahiwal cattle further emphasizes the role of long-term selection pressure and adaptation in shaping the genetic makeup of these breeds. Selection sweeps represent genomic regions where specific alleles have been favored and enriched over generations due to their beneficial effects on fitness or productivity traits (Illa et al., 2021). This indicates ongoing selection processes within these breeds, possibly driven by environmental pressures or breeding objectives.
The presence of candidate genes within these selection sweep regions suggests their potential role in adaptation to local environments, disease resistance, or other fitness-related traits. Thus, understanding the genetic basis of selection sweeps can provide valuable insights into the evolutionary history and adaptive potential of livestock populations.
GO and pathway analysis of genetic diversity and selection sweep regions in Sahiwal cattle
GO enrichment analysis is a powerful bioinformatics tool used to identify biological processes, cellular components, and molecular functions that are significantly overrepresented within a given set of genes. In the context of genome-wide genetic diversity regions in Sahiwal cattle, this analysis can provide insights into the functional implications of genetic variation. This study utilized GO enrichment analysis to identify key biological processes, cellular components, and molecular functions associated with genes located in these regions, thereby shedding light on the potential adaptive and functional roles of these genes in Sahiwal cattle (Figs. 1 and 3).
In Supplementary Table S4, the GO enrichment analysis of biological processes revealed several significant categories. The significant enrichment of genes related to “axon guidance” (GO:0007411), including EFNA1, DSCAM, SLIT1, UNC5C, and SLIT3, with a fold enrichment of 8.41, suggests that genetic diversity in Sahiwal cattle may influence neural development pathways, particularly axonal growth, which could affect behavioral traits and responses to environmental stimuli (Chicherova et al., 2023; Hao et al., 2021; Montesinos, 2014). Additionally, the enrichment of genes involved in “modulation of synaptic transmission” (GO:0050804), such as LRFN2, PLCB4, and NRG3, with a fold enrichment of 13.20, implies that synaptic plasticity and neurotransmission are important processes in this breed, potentially impacting learning, memory, and adaptability to environmental changes (Chen et al., 2022; Falls, 2003; Muller et al., 2018; Zhou et al., 2022). For instance, LRFN2 is linked to hippocampal long-term potentiation and synaptic activity through AMPA receptor regulation (McMillan et al., 2021), which may enhance cognitive flexibility in Sahiwal cattle, allowing them to better adapt to environmental and management challenges. This genetic diversity likely contributes to their resilience, stress tolerance, and behavioral traits that are advantageous for thriving in tropical climates.
Other notable enrichment of genes involved in “axon extension involved in axon guidance” (GO:0048846) and “protein dephosphorylation” (GO:0006470) in Sahiwal cattle underscores the critical role of neural development and signal transduction pathways in this breed. Genes such as SLIT1, SLIT3, PTPRD, PPP2R1A, and PPM1H are prominently featured, suggesting that genetic diversity in these processes may significantly impact the cattle’s ability to handle various environmental and physiological challenges (Berndsen et al., 2019; Jiang et al., 2019; Uhl and Martinez, 2019; Wang et al., 2023). Specifically, SLIT3 has been shown to inhibit normal corneal epithelial injury repair and nerve regeneration, as well as significantly suppress the proliferation and migration of cultured mouse corneal epithelial cells (Chen et al., 2024). This indicates that genetic variations in SLIT3 and other related genes could influence not only neural development but also the cattle’s capacity for adaptation and recovery from injuries or stressors, highlighting their potential role in enhancing resilience and managing environmental stressors.
Supplementary Table S5 highlights significant findings from the GO enrichment analysis of cellular components, particularly noting a substantial enrichment in the “dendrite” category (GO:0030425) with a fold enrichment of 6.59. This suggests that genes involved in dendritic structure and function, such as C4A, HSPA8, PLCB4, DSCAM, PPP2R1A, and UNC5C, are critical contributors to the genetic diversity observed in Sahiwal cattle. The “neuronal cell body” (GO:0043025) category was also significantly enriched, with genes like GRIA1, DSCAM, PPP2R1A, UNC5C, and ASIC2 contributing to this process. This enrichment underscores the vital role of dendritic architecture in neural connectivity and signal processing, which may impact cognitive and sensory functions (Dewa et al., 2024; Hizawa et al., 2024; Sasai et al., 2021; Stricher et al., 2013; Treccarichi et al., 2024; Wang and Liu, 2021; Wang et al., 2023).
Supplementary Table S5 also displays significant enrichment in the “glutamatergic synapse” (GO:0098978) and “Golgi apparatus” (GO:0005794) categories. The enrichment of genes involved in synaptic transmission, such as PTPRD, GRIA1, PLCB4, NRG3, PPM1H, and PRKN, suggests that these genes play a pivotal role in the genetic diversity of Sahiwal cattle. These genes are essential for glutamatergic synaptic signaling and protein processing within the Golgi apparatus, which are critical for maintaining neural function and cellular homeostasis. The involvement of these genes in synaptic signaling processes underscores their potential influence on neural connectivity and cognitive functions (Ang et al., 2021; Cortés et al., 2024; Muller et al., 2018; Uhl and Martinez, 2019; Watzlawik et al., 2024; Wu et al., 2019). Moreover, their role in intracellular transport and protein processing within the Golgi apparatus highlights their importance in cellular function and overall adaptability in Sahiwal cattle. This genetic enrichment indicates that these pathways could be critical for the breed’s neural and cellular adaptability, influencing various aspects of its health and performance.
The GO enrichment analysis of molecular functions, as presented in Supplementary Table S6, highlights significant findings in categories related to calcium ion binding (GO:0005509), motor activity (GO:0003774), and actin binding (GO:0003779). The calcium ion binding category, with a fold enrichment of 3.90, includes genes such as CRACR2A, PLCB4, CAPNS1, CRTAC1, KCNIP3, SLIT1, TLL2, SLIT3, and ASTN2. This suggests that calcium signaling pathways, crucial for muscle contraction, neurotransmission, and signal transduction, may be significantly influenced by the genetic diversity in Sahiwal cattle. Earlier studies also demonstrated that regions associated with calcium channels, including CACNA1S and CRACR2A, are important for regulating calcium levels in Holstein cows (Cavani et al., 2022). Specifically, CRACR2A facilitates calcium ion entry through the cell plasma membrane and interacts with ORAI1 to control intracellular calcium levels (Lopez de Heredia et al., 2020). Additionally, the cows with subclinical hypocalcemia showed reduced mRNA expression of ORAI1 in neutrophils, which likely impairs calcium ion entry during cell activation (Zhang et al., 2019). These findings underscore the pivotal role of calcium signaling pathways in cellular function and suggest that genetic variation in these pathways may significantly impact physiological processes in Sahiwal cattle.
The enrichment of genes involved in motor activity, such as KIF26B, MYO18B, and MYO16, with a notable fold enrichment of 24.25, underscores their significant role in cytoskeletal dynamics and cellular movement. This enrichment suggests that these genes are crucial for muscle development and function in Sahiwal cattle, potentially influencing their physical adaptability and performance traits. In similar study, transcriptomic gene profiling in porcine muscle tissue has provided insights into histological properties and interactions between actin filaments and membrane transport proteins. Genes such as ACTC1, TNNT2, and the myosins MYO18B and MYO16 exhibited divergent expression levels, potentially influencing muscle fiber size and overall muscle characteristics across different muscles (Molik et al., 2017). This underscores the role of these motor activity genes in muscle development and function, ultimately contributing to the physical attributes and performance capabilities of Sahiwal cattle.
In addition to analyzing genetic diversity regions, GO and pathway analyses (Figs. 2 and 4) were conducted for genes identified in selection sweep regions in Sahiwal cattle (Supplementary Tables S7, S8 and S9). The analysis reveals that genes involved in the regulation of membrane potential and ion transmembrane transport, such as RIMS1, TAFA4, ASIC2, and KCNH1, are crucial for maintaining cellular ionic balance and facilitating ion movement across membranes, which is essential for cellular homeostasis and stress response (Hoeffel et al., 2021; Napoli et al., 2022; Sivils et al., 2022; Wang et al., 2018). Additionally, genes like COLEC12 and CAV1, associated with the positive regulation of cell adhesion molecule production, are important for cell-cell interactions and tissue integrity. The negative regulation of nitric-oxide synthase activity by CAV1 and ATP2B4 highlights their role in modulating nitric-oxide levels, which are critical for cellular signaling and immune responses. The analysis also identifies enriched processes related to response to stimuli and interleukin-15-mediated signaling, emphasizing their roles in environmental response and immune regulation. Similar, studies reported that the annotation and genetic diversity of chicken collagenous lectins, including COLEC12, and performed a comparative genomic analysis using publicly available data (Hamzic et al., 2015). Their findings revealed that collectins and ficolins, especially COLEC10, COLEC11, and COLEC12, have conserved protein sequences and gene structures across all vertebrate groups, highlighting the evolutionary significance of these genes.
The observed enrichments in various cellular components reveal critical aspects of cellular function and stability. The presynaptic membrane, enriched with genes like RIMS1, KCNJ3, and KCNH1, is pivotal for neurotransmission and synaptic signaling (Tian et al., 2023; Wang et al., 2018; Yamada et al., 2019). Similarly, the membrane and cytosol, featuring genes such as SLC45A4, COLEC12, RIMS1, and KSR1, emphasize their roles in maintaining cellular structure and function (Chen et al., 2021; Li et al., 2020; Liu et al., 2023). The integral component of the plasma membrane, represented by genes such as CD4, GPA33, CAV1, and ATP2B4, is crucial for sustaining cellular integrity and external interactions (Erdogmus et al., 2022; Hattangady et al., 2020; Merino-Wong et al., 2021; Opstelten et al., 2021). Additionally, the Z disc component, with genes such as PDLIM1, ATP2B4, and CRYAB, is associated with muscle function and cell stability, while the cell cortex and adherens junctions, featuring genes like CLIC5 and PARD3, are essential for cellular architecture and adhesion. These findings underscore the complex interplay between cellular components and their roles in maintaining cellular homeostasis and function.
Molecular function analysis reveals significant enrichments in several key areas, highlighting their roles in cellular activities and stability. The enrichment in metal ion binding, with genes such as COLEC12, PRIM2, ATP2B4, and HDAC9, underscores their importance in enzymatic functions and cellular stability (Li et al., 2020; Yang et al., 2021). ATP-binding genes, including UCK2, KSR1, and SYK, are crucial for energy metabolism and signaling processes (Cai et al., 2020). Additionally, genes associated with nitric-oxide synthase binding and transcription coactivator activity play significant roles in gene regulation and signaling pathways. The enrichment of GTPase activator activity and protein binding, particularly in cell-cell adhesion, further highlights the genes roles in cellular communication and structural stability. Collectively, these findings provide valuable insights into the genetic basis underlying adaptive traits and selection pressures in Sahiwal cattle.
The pathway analyses of identified genes in genome-wide genetic diversity and selection sweep regions of Sahiwal cattle, presented in Supplementary Tables S10 and S11, reveal important insights into the biological processes potentially under selection in Sahiwal cattle. Genes identified in regions associated with genome-wide genetic diversity are significantly enriched in pathways related to axon guidance, long-term depression, and glutamatergic synapse (omi.2024.0188_Supplementary_Table_S10). These pathways are crucial for neuronal development and synaptic plasticity, indicating a potential link between genetic diversity in Sahiwal cattle and traits related to the nervous system. Furthermore, pathways associated with glycosaminoglycan biosynthesis, including chondroitin sulfate/dermatan sulfate and heparan sulfate/heparin, are also highlighted. Genes involved in these pathways, such as CHST11, XYLT1, and HS6ST3, suggest important roles in connective tissue development and maintenance, which could be significant for structural integrity and resilience (Lin et al., 2023; Mis et al., 2014; Zhao et al., 2015). These findings suggest that genetic diversity in Sahiwal cattle may impact both neuronal and connective tissue-related traits.
The analysis of genes identified in genome-wide selection sweeps regions, as detailed in Supplementary Table S10, shows significant enrichment in several pathways, including the phospholipase D signaling pathway, bacterial invasion of epithelial cells, and aldosterone synthesis and secretion. The phospholipase D signaling pathway, involving genes such as SYK, PLCB1, and CYTH1, is essential for intracellular signaling and membrane dynamics, which could be linked to immune responses and cellular communication (González-Burguera et al., 2024; Ren et al., 2024). The pathway for bacterial invasion of epithelial cells, with genes such as ARHGAP10 and CAV1, suggests an adaptive immune response that may have been selected to enhance disease resistance in Sahiwal cattle (Imam et al., 2020). Additionally, the enrichment in the aldosterone synthesis pathway, featuring genes such as CREB3L2 and ATP2B4, points to potential selection for traits related to water and electrolyte balance, which could be crucial for adaptation to heat stress and arid conditions.
Overall, these findings provide a deeper understanding of the genetic factors that contribute to the adaptability and resilience of Sahiwal cattle, emphasizing the importance of both genetic diversity and selective pressures in shaping the breed’s characteristics.
Conclusions
The present study identified 728 selection sweeps across the Sahiwal cattle genome, with notable concentrations on chromosomes 6, 8, and 10, indicating strong genetic adaptation and selective pressures. These sweeps encompass 89 genes associated with key traits such as adaptability, body linear traits, production, reproduction, and disease resistance. The identified genes are involved in essential pathways, including gluconeogenesis, lipid metabolism, and response to stimuli, influencing physiological functions crucial for adaptability and disease resistance. Notably, the genetic adaptations observed reflect the breed’s ability to respond to environmental challenges, including those driven by climate change, underscoring their resilience and adaptive capacity. This genetic evidence highlights the breed’s significant evolutionary adjustments driven by selective forces, enhancing their resilience and adaptability to changing environmental conditions. Although the study was conducted on a relatively small sample size, the findings provide a valuable foundation for future marker-based validation studies, offering insights into the genetic basis of adaptability and selective advantages in Sahiwal cattle under the pressures of climate change.
Footnotes
Authors’ Contributions
Data curation and methodology: A.M. and P.C. Formal statistical data analysis: A.M. and Y.C.B. Overall conceptualization: A.M. Wet lab work/laboratory work assistance: P.S., P.C., and A.R.G. Bioinformatics software analysis: R.A. Writing—original draft: A.M., M.K.R., P.C., and P.S.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
Funding Information
No funding was received for this article.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.