
Abstract
Interleukin-2-inducible T-cell kinase (ITK) is a critical tyrosine kinase enzyme that is involved in the activation and differentiation of T cells. ITK is mainly found in T cells, which plays an essential role in controlling T-cell receptor signaling and downstream pathways. ITK regulates the synthesis of cytokines, particularly interleukin-2 (IL-2), and the development of Th2 cells. ITK is of interest for drug discovery and molecular targeting in immunology, autoimmune diseases, and cancer. Here, we report a structure-based virtual screening utilizing a collection of small molecules obtained from the PubChem database with an eye on the discovery of drugs targeting ITK. The compounds were selected according to compliance with the Lipinski’s rule of five. The molecular docking investigation focused on prioritizing binding affinity and specific interaction toward the kinase domain. The highest-ranking search results were subjected to identification of possible pan-assay interference compounds (PAINS), assessment of pharmacokinetic properties, and estimation of pharmacological activity using Prediction of Activity Spectra of Substances (PASS) analysis. The interactions among these chemicals and the salient residues in the interleukin-2-inducible T-cell kinase (ITK) kinase domain were unpacked using a two-dimensional approach. The reference inhibitor ITK-Inhibitor-2 (IMM) and four elucidated compounds with PubChem CIDs, namely, 90442621 (PFB), 141764004 (FTP), 149213796 (FPP), and 145983307 (MBD), showed significant binding affinity of −8, −10.4, −9.8, −10.2, and −10.7 kcal/mol, respectively, and high selectivity for the ITK binding pocket. In conclusion, this study reports on the potential of several compounds for therapeutic targeting of ITK. Furthermore, structural analysis revealed the interaction of proposed compounds and active site residues within the ATP-binding pocket is highly similar to known inhibitors but shares distinct interaction patterns that could improve specificity. This specificity and optimization hold potential for the development of next-generation ITK inhibitors with possible applications in the treatment of immune-related disorders and cancers. Further in vitro, in vivo, and translational clinical research are called for.
Introduction
Interleukin-2-inducible T cell kinase (ITK) is an essential member of the Tec family of nonreceptor tyrosine kinases, playing a pivotal role in T-cell activation and signaling. It functions through antigen receptors, including the T-cell receptor and B-cell receptor, which regulate immune responses. ITK is particularly involved in modulating Th2, Th9, and Th17 cell responses, thereby influencing the production of pro-inflammatory cytokines (Brown et al., 2004). ITK was discovered in 1993 by two different research groups led by Gary Koretzky and Arthur Weiss (Janeway and Bottomly, 1994). The native form of the ITK protein is 620 amino acids long with a molecular weight of 71,831 Da. In humans, it is positioned at the chromosomal location 5q33.3. It is classified as a transferase inhibitor and a crucial component of T cell signal transduction, as well as plays a significant role in proliferation, cytokine release, and chemotaxis. It comprises of several functional domains, which includes the Tec homology (TH) domain, pleckstrin homology (PH) domain, Src homology 2 (SH2) domain, Src homology 3 (SH3) domain, and Src homology 1 (SH1), also known as kinase domain (Brown et al., 2004; Yang et al., 2000).
The kinase domain has one ATP binding site on Lys391, an active site on Asp482, and a nucleotide binding site Ile-Gly-Ser-Gly-Glu-Phe-Gly-Leu-Val. The PH domain of ITK is located at its amino-terminal and functions to bind phosphorylated lipids on membranes, and the proline-rich region (PRR) of the TH domain is required for binding to the SH3 domain (Andreotti et al., 1997; Hao and August, 2002). A binding interaction occurs between the SH3 domain and the PRR in the TH domain, which results in the auto-inhibition of ITK whereas PLC-1 is bound by the SH2 domain, which permits protein–protein interactions (Ching et al., 2000; Pawson et al., 2001). To simplify, mutations or deletions in the SH3 domain result in the activation of ITK, whereas mutations and deletions in the SH2 domain led to its inactivation. An ATP binding pocket is located at the carboxyl end of ITK, within the SH1 domain, where the kinase activity of ITK is occurred (Heyeck et al., 1997). This leads to the downstream activation of multiple signaling pathways essential for T-cell functioning. ITK is also engaged in the control of cytokine production, particularly IL 2, and in the differentiation of TH2 cells (Bunnell et al., 2000; Fowell et al., 1999).
The expression of ITK is primarily found in healthy T cells, particularly in CD4+ T helper cells and T-cell related hematological malignancies. Its essential role in regulating T lymphocyte activity in immune-mediated disorders and lymphoproliferative diseases induced by EBV has been well-known (Andreotti et al., 2010; Huck et al., 2009). A wide range of conditions known as lymphoproliferative disorders (LPDs) (Chen et al., 2019; Sawada and Inoue, 2018) are distinguished by excessive lymphocyte proliferation that causes bone marrow invasion, lymphadenopathy, and monoclonal lymphocytosis (CVID). Patients undergoing tissue transplantation and those on immunosuppressive medications, including cyclosporin, sirolimus, and tacrolimus, are also at risk. This pathology has also been associated with invasive fungal infections (Justiz Vaillant and Stang, 2024). An uncontrolled proliferation of T and B cells, one type of lymphocyte, can cause immune-proliferative diseases. These can cause immunodeficiency, a weak immune system, and problems with the management of lymphocytes (Al-Saleem and Al-Mondhiry, 2005). Epstein Barr virus (EBV) is the primary cause of most LPDs. It likely transmits by saliva droplets. It takes between 4 and 5 weeks for the incubation process to complete.
The treatment for primary hemophagocytic lymphohistiocytosis, an EBV T/NK cell lymphoproliferation, may involve the administration of steroids, cyclosporine A, and etoposide. Certain individuals are known to have remission; however, to achieve this, chemotherapy may be required (Sawada and Inoue, 2018). Three deadly outcomes can result from the X-linked lymphoproliferative syndrome, which is marked by an incorrect immunological reaction to EBV. These outcomes are infectious mononucleosis, dysgammaglobulinemia, and B-cell lymphoproliferative diseases. However, there is no information available on the connection between EBV and X-linked lymphoproliferative illness (NCBI, 2025). Several research works on ITK inhibition have already been performed, and many are still going on. Because of its central role in immune responses, ITK is involved in various immune-related conditions and certain cancers. Therefore, our objective was to discover small molecule inhibitors that can selectively target ITK. Combining structure-based drug designing (SBDD) with virtual screening (VS) can be a strategy for drug development (Araujo et al., 2013; Lionta et al., 2014). SBDD employs precise structural knowledge to direct the design process, and VS effectively evaluates extensive chemical libraries to forecast possible strong receptor binding (Naqvi et al., 2018).
In our study, we prepared a library from similar structures with a Tanimoto threshold of 80% of the existing ITK inhibitors where 2649 compounds were identified using Lipinski’s rule of five from PubChem. We performed VS on these compounds against ITK using InstaDock software suite (Mohammad et al., 2021) to discover high-affinity lead compounds. The top hits were chosen according to their strong binding affinity and fundamental characteristics, followed by ADMET analysis (Pires et al., 2015). Molecules that formed direct bonds with the ITK binding site through interactions were subsequently selected. In addition, molecular dynamics (MD) simulation was conducted, thereby replicating the atomic motion and behavior of molecules over a period, offering a valuable understanding of the stability and dynamics of the system. Supplementary Figure S1 illustrates the systematic representation of the methodologies utilized in this study.
Material and Methods
The present study used computational biology methods and did not require research ethics board approval. The study was conducted under the overall research ethics oversight of the authors’ institutions.
Computational resources and tools
This study was performed by employing a workstation with a 3.47 GHz 8-core CPU and a Nvidia 4080 16 GB GPU. Although performing this experiment, various online resources were utilized for the retrieval of data and analysis, including UniProt (Consortium, 2020), RCSB Protein Data Bank (PDB) (Berman et al., 2000), SwissADME (Daina et al., 2017), PASS online server (Filimonov et al., 2014), pkCSM (Pires et al., 2015), and IMPPAT database (Vivek-Ananth et al., 2023). Furthermore, a few stand-alone bioinformatics programs were employed, including PyMOL (Delano, 2002), QtGrace, InstaDock v1.2, and Discovery Studio Visualizer (Biovia DS, 2017).
Receptor and ligand preparation
Using UniProt and PDB, the appropriate ITK structure was identified and retrieved. It was selected because of its high resolution, presence of kinase domain, ligand association, and structural purity. The ITK protein of Homo sapiens was retrieved from RCSB in PDB format (PDB ID: 4M15, Resolution: 1.52 Å). PyMOL (Delano, 2002) was then used to further process the structure by removing co-crystal ligands, water molecules, and heteroatoms.
Database for screening
Constructing a compound library with a diversified structural representation in the database is essential for developing an effective VS method. This diversity enhances the chances of discovering potential hits with therapeutic value. Several freely accessible databases, including PubChem, DrugBank, the National Cancer Institute database, and the Binding Database, provide invaluable resources for assembling small molecule libraries. The colibrary utilized here was prepared from similar structures of existing inhibitors. The initial compound library was constructed using the structural frameworks of six known ITK inhibitors to maximize chemical diversity and ensure comprehensive scaffold representation. This co-library contains 6000 bioactive molecules, of which 2649 compounds were combined into a library using the Lipinski’s rule of five. It defines precise boundaries for important physicochemical characteristics that are essential to a compound’s potential use as a drug molecule. While constructing the library, compounds that did not satisfy any of these requirements were eliminated from consideration (Mohanraj et al., 2018). After docking the filtered library and analyzing binding affinities, the selected hits were structurally similar to only four inhibitors: ITK-inhibitor-2, ITK-TRKA-IN-1, PF-06465469, and PRN 694. Consequently, these four inhibitors were used as reference molecules in subsequent analyses to maintain relevance to the high-affinity compounds identified.
Input files and directories
Here, we utilized InstaDock, an easy-to-use Windows-based graphical software for molecular docking-based VS (Mohammad et al., 2021). The initial step in molecular docking analysis is to create an organized working directory that includes the ligand files, the receptor PDB file, and InstaDock. By transforming the chemical format into an appropriate PDBQT format and assuring compatibility for docking tasks, InstaDock optimizes the process. Additionally, to ensure accurate and regulated docking conditions, a configuration file is created that specifies the 3D space in which all the ligands will be engaged in docking.
Molecular docking screening methodology
With the goal of screening ligands against the ITK crystal structure, the receptor–ligand docking approach was applied. Understanding the position of ligands within a receptor’s active site cavity is essential for understanding the target-inhibitor selectivity process, and molecular docking plays a vital part in this process (Mohammad et al., 2020). A grid box was constructed during the docking process to define a precise docking region that ensures an optimal binding affinity and structural placement of ligand-protein complexes in the space. The dimensions of the grid box were specified by InstaDock in this context, with the size parameters (X, Y, Z) as 61, 66, and 68, and the center coordinates (x, y, z) being 5.147, 2.718, and 10.824. Using the docking approach, various ligand orientations within the cavity of the receptor’s active site were investigated. The scoring function generated by each ligand docking serves as the foundation for determining the ligand’s binding affinity to the protein (Meng et al., 1992). Following the ligand screening process, InstaDock read data from the log files and out files to evaluate the docking results. The most optimal docked conformation was then chosen for further study.
We used several computational methods to evaluate the characteristics and potential therapeutic candidates among the filtered drugs. We assessed the presence of PAINS patterns by utilizing the SwissADME server and then analyzed their physicochemical properties, comprising characteristics linked to toxicity, distribution, metabolism, excretion, and absorption using the pkCSM server. Further, the PASS tool was utilized to assess the pharmacological characteristics of the interactions between ligands and the receptor, with an emphasis on those that have a probability (Pa) that exceeds 0.7. Through these thorough evaluations, we were able to acquire a deep understanding of the potential of the compounds and their suitability for future therapeutic development (Han et al., 2019). The aim for assessing PAINS was to eliminate the false positive compounds to increase the reliability of findings, whereas certain thresholds for pharmacokinetic properties were set to ensure the compounds were in compliance with Lipinski’s RO5.
Molecular dynamics simulations
The VS process led to molecular mechanics-level analysis, where a 300 ns MD simulation was conducted using the CHARMM36 force field. This simulation, performed at 300K in the GROMACS suite, included three selected compounds along with unbound ITK to evaluate their stability and interactions (Qi et al., 2012; Sahu and August, 2009; Siveen et al., 2018). The CGenFF server was utilized to generate force field parameters and topologies for each compound. The ligand–ITK complexes were solvated within a 10 Å cubic box using the gmx solvate module, employing the TIP3P water model. To ensure system stability, energy minimization was conducted through the steepest descent approach. The equilibration process was executed under periodic boundary conditions, where the system temperature was progressively raised from 0K to 300K over a 1000 ps interval at a constant volume. MD trajectories were analyzed to assess structural and dynamic properties using various time-evolution metrices. Graphs and visualizations illustrating ITK residual interactions and stability postligand binding were generated using QtGrace.
MMPBSA calculation
The Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) method is a widely utilized computational approach for estimating the binding free energy between a protein and a ligand. The open-source program gmx_MMPBSA was used to estimate the binding free energy of ITK and its bound ligands (Valdés-Tresanco et al., 2021). A time scale of 10 ns is included in the trimmed stable trajectory, with each frame taken at every 10 ps. This is a tool created to execute free energy calculations on trajectories generated by GROMACS simulations. The binding energy estimation in this study considered multiple components, including the system’s internal energy, the electrostatic free energy of solvation, and the nonpolar solvation energy. The binding free energy (ΔGbinding) of the ligand–protein complexes was computed by:
Where the total binding energy of the protein-ligand complex, denoted as
Result and Discussion
Computational analysis of docking results
The library was constructed using the structural framework of six known inhibitors of ITK with a Tanimoto similarity index threshold of ≥80% to ensure a comprehensive representation of diverse binding scaffolds. These inhibitors were selected based on their IC50 values. The reference inhibitors, along with their IC50 values, are given in Supplementary Table S1. After docking, we found that the top nine compounds with the best binding affinities align structurally with only four of these six inhibitors. We identified the most representative molecule from the selected inhibitors based on its binding affinity and pharmacological relevance, making it a suitable candidate for comparative studies with the selected hits. These data provide a benchmark for comparing the identified compounds’ predicted affinities and pharmacokinetic properties, highlighting their potential as potential next-generation ITK inhibitors. Library contains a total of 6,000 compounds, out of which 2649 compounds were selected according to the pharmacological similarities, complying with the Lipinski’s rule of five. An extensive approach to the VS of ligands was implemented to identify inhibitors targeting the ITK binding site. The docking-generated outputs were subsequently utilized to exclude compounds with unfavorable binding affinities.
The docking process validation results demonstrated that the protocol reliably reproduced the binding pose of ADP as observed in the ITK co-crystal structure (PDB ID: 4M15). This structure was chosen due to the higher resolution (1.52 Å) and no mutational occurrence. The overlay of the docked and cocrystallized poses highlighted their alignment, confirming the protocol’s accuracy (Supplementary Fig. S2). The docking results for the co-crystallized ligand QWS showed a binding affinity of −8.4 kcal/mol and a ligand efficiency of 0.32 kcal/mol/heavy atom. After filtering the docked result, it was found that 60 out of 2649 molecules had a considerable binding affinity to ITK, ranging from −10.8 to −9.3 Kcal/mol (Supplementary Table S2). Further, ADMET analysis of these compounds resulted in the selection of 9 potential small molecules with inhibitory properties against ITK. Table 1 displays the docking score of the nine hits along with the known inhibitors.
Comparison of the Chosen Compounds with Four Reference Molecules, i.e., ITK-Inhibitor-2, ITK-TRKA-IN-1, PF-06465469, PRN 694, and Co-Crystallized Ligand
Ligand efficiency values are in Kcal/Mol/heavy atom.
ITK, interleukin-2-inducible T-cell kinase.
Pharmacokinetic evaluation
The pharmacokinetic properties of a ligand can help predict its potential as a therapeutic candidate and its chances of success in clinical trials. The findings demonstrate that each molecule satisfied Lipinski’s rules, an essential formula for estimating the possibility of a medicine. To assess the PAINS filter and ADMET characteristics of the chosen hits from the docking study (Consortium, 2020), we employed SwissADME and pkCSM. We have identified nine compounds with favorable pharmacokinetic and physicochemical properties. They might be more effective drug candidates because they might not contain any harmful properties (Supplementary Table S3). Table 2 lists the ADMET evaluation of the chosen hit molecules. The findings suggest that the nine compounds exhibit promising physicochemical characteristics and lack any PAINS patterns, making them potential candidates for further therapeutic advancement.
ADMET Properties of the Nine Chosen Compounds and the Four Known Inhibitors
PASS analysis
PASS is a highly useful tool for evaluating the biological capabilities of a small chemical compound. PASS predicts various sorts of biological activities according to the structure of the chemical (Lagunin et al., 2000). Using SMILE strings through the Way2Drug PASS Online platform, all nine chosen compounds were analyzed. The purpose is to identify ligands that possess anticancer and autoimmune disease properties, with the goal of finding a potential therapeutic option for ITK-mediated diseases. After careful study, four chemical compounds with PubChem CIDs: 90442621 (PFB, N-[4-methyl-3-[8-[(3R)−3-methylmorpholin-4-yl]imidazo[1,2-a]pyrazin-6-yl]phenyl]−3-(trifluoromethyl)benzamide), 141764004 (FTP, N′-[2-fluoro-5-(trifluoromethyl)phenyl]-N-(3-pyridin-3-yl-1H-indazol-6-yl)propanediamide), 145983307 (MBD, 8-(2-methyl-3H-benzimidazole-5-carbonyl)−2-[4-(trifluoromethyl) phenyl]−2,8-diazaspiro [4.5] decan-3-one), and 149213796 (FPP, N-[5-[3-[4-(3-fluoro-5-methylphenyl)−1H-benzimidazol-2-yl]−1H-pyrazolo[4,3-b]pyridin-5-yl]pyridin-3-yl]propanamide) were chosen based on their biological activities. The activity and Pa and Pi values of the four chosen compounds are presented in Supplementary Table S4. The reference inhibitor ITK-inhibitor-2 (IMM) also showed anticancer properties, which validate the PASS predictions.
Interaction analysis
Protein–ligand interaction analysis is essential in drug discovery, as it helps assess how small molecules (ligands) interact and bind to target proteins. The 3D interaction of ITK with the reference molecule, IMM, is illustrated in Supplementary Figure S1. The docked conformations were evaluated by examining interaction residues. The analysis revealed that all four molecules exhibit specific interactions with crucial amino acid residues of the ITK binding site (Fig. 1). IMM (ITK-inhibitor-2) was selected as the reference molecule due to its well-characterized binding interactions and high selectivity for ITK. Among the six inhibitors used in the library construction, IMM demonstrated the highest pharmacological relevance and binding affinity. Its selection ensures a robust comparative framework for evaluating the identified compounds.

ITK residual interaction with selected compounds.
MD Simulation studies
Following the VS procedure, we carried out MD simulations to examine the structural dynamics and stability of ITK and its ligand-bound systems. Four selected hits from the library and one reference molecule were included in these simulations, which were performed on the docked complexes in predefined solvent conditions. For each 300 ns simulation, the selected compounds’ initial orientations served as the starting positions. Various structural properties and characteristics were analyzed throughout the simulations. This comprehensive study provided insights into the dynamic behavior of ITK and its ligand-bound systems.
The root mean square deviation (RMSD) analysis was performed to study free ITK structure and the receptor-ligand complexes, including ITK-IMM, ITK-PFB, ITK-FTP, ITK-FPP, and ITK-MBD. Supplementary Table S4 shows the RMSD values of all six systems. Figure 2 presents the structural changes in ITK upon interaction with IMM, PFB, FTP, FPP, and MBD, revealing minor but consistent fluctuations. Notably, the ITK-IMM and ITK-PFB complexes exhibited greater stability, reflected in lower RMSD values compared to the ITK and the other three complexes. The RMSD plot in Figure 2A indicates that ITK-IMM and ITK-PFB complexes experienced fewer fluctuations than the unbound ITK. The root mean square fluctuation (RMSF) analysis showed that the backbone of all four systems ITK, ITK-IMM, and the ITK-PFB, ITK-FTP, ITK-FPP, and ITK-MBD complexes, exhibited RMSF values with random peaks. The average RMSF values observed were 0.08 nm for ITK, 0.07 nm for ITK-IMM, and 0.08 nm, 0.08 nm, and 0.09 nm for the ITK-PFB, ITK-FTP, ITK-FPP, and ITK-MBD complexes, respectively. As shown in Figure 2B, the compounds displayed numerous steady fluctuations throughout the trajectory. However, beyond amino acid residue 520, all three complexes demonstrated a stable and similar RMSF patterns.
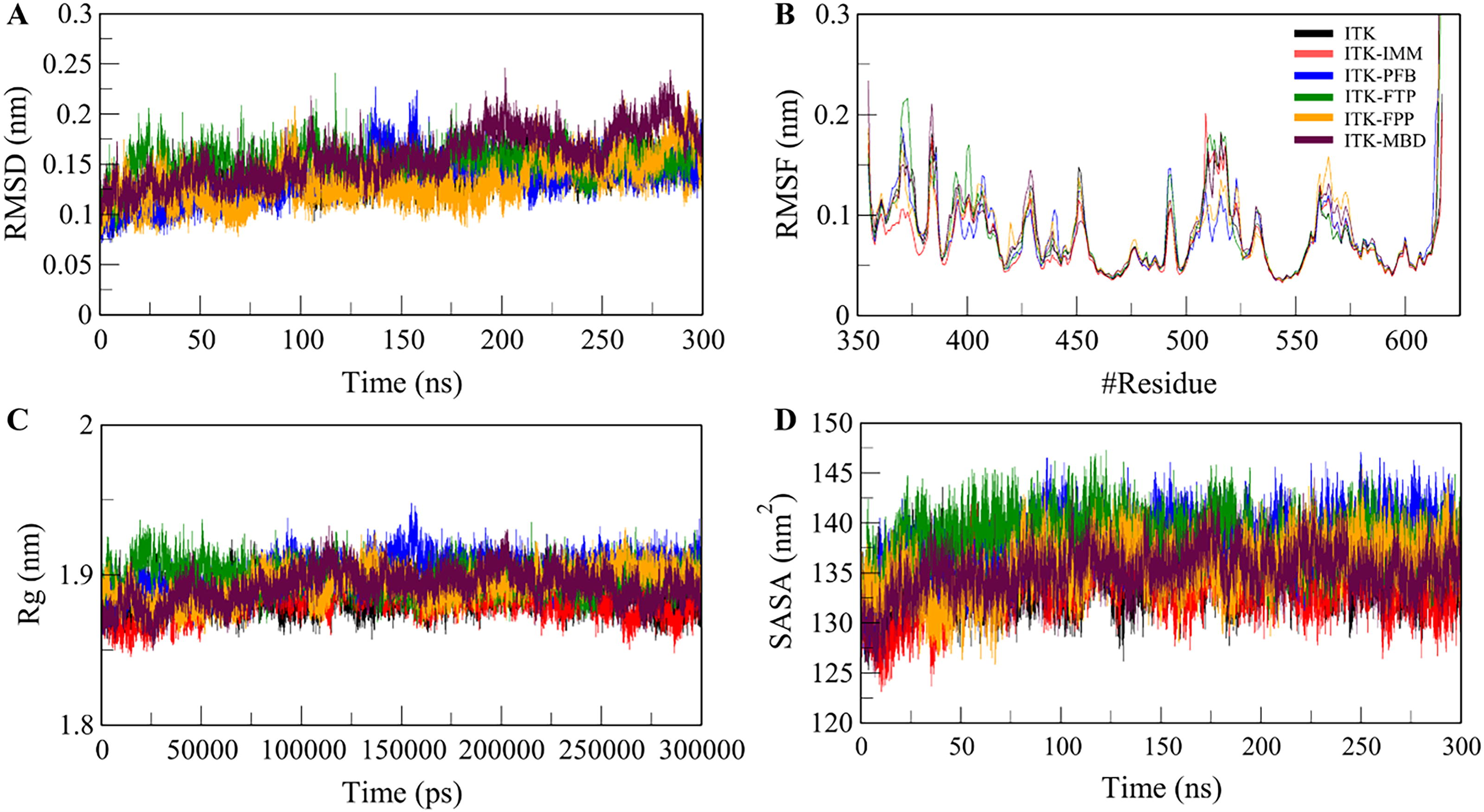
Structural dynamics and compactness of ITK upon binding with the four selected compounds PFB, FTP, FPP, MBD and reference molecule IMM
The radius of gyration (Rg) provides statistical insights into the protein folding within biological systems (Anjum et al., 2022). Rg is commonly used to assess the compactness of a protein structure. The average Rg for ITK, reference molecule ITK-IMM, and the four complexes, ITK-PFB, ITK-FTP, ITK-FPP, and ITK-MBD, displayed a consistent degree of uniformity. Figure 2C illustrates the time evolution of Rg, showing that all complexes remained stable with well-maintained structural integrity and folding, leading to a minimized Rg. Solvent accessible surface area (SASA) measures the extent of a protein’s surface that interacts with the surrounding solvent (Ali et al., 2014; Mazola et al., 2015). Figure 2D presents the SASA plot, where the average SASA were calculated for ITK and its complexes. The analysis indicated slight fluctuations in SASA throughout the simulations. The average SASA for ITK, ITK-IMM and the four complexes ITK-PFB, ITK-FTP, ITK-FPP, and ITK-MBD were 134.5, 133.8, 138, 138.4, 135.5, and 135 nm2, respectively (Supplementary Table S4).
Dynamics of hydrogen bonds
Hydrogen bond formation plays a vital role in the conformational dynamics of proteins and their ligand-bound complexes. We evaluated the intramolecular hydrogen bonds in both the unbound ITK structure and ITK complexes with IMM, PFB, FTP, FPP, and MBD. Graphs were generated over 300 ns to evaluate the folding kinetics across the six systems (Supplementary Fig. S5). The plots indicate that the number of intramolecular hydrogen bonds remained relatively consistent among the unbound protein, the reference molecule complex, and the four ligand-bound complexes. As shown in Supplementary Table S5, the average hydrogen bonds before and after complex formation with IMM, PFB, FTP, FPP, and MBD were 188, 188, 184, 183, 191, and 187, respectively. This suggests that ligand binding did not induce any major change in the overall hydrogen bonding pattern of the protein with the solvent. Additionally, a probability density function (PDF) graph depicting intramolecular hydrogen bonds confirmed their stability and reliability across the simulations (Supplementary Fig. S5B). These findings indicate that intramolecular hydrogen bonds within ITK remained stable throughout the simulations in all six systems.
We also investigated the dynamics of intermolecular hydrogen bonding between ITK and the selected small molecules. All complexes demonstrated stable hydrogen bonding interactions, with the ITK-FPP complex exhibiting the highest number of hydrogen bonds in all the systems. Specifically, ITK-FPP consistently maintained 3–4 hydrogen bonds during the whole trajectory (Fig. 3A). In contrast, the other four complexes, ITK-IMM, ITK-PFB, ITK-FTP, and ITK-MBD, retained stable 2–3 hydrogen bonds across the simulation period (Fig. 3). These results indicate that the protein-ligand complexes’ overall structural integrity remained unchanged over time. The ability of the complexes to maintain their docking positions was strengthened by the persistence of intermolecular hydrogen bonds, suggesting strong and stable binding interactions throughout the simulation.
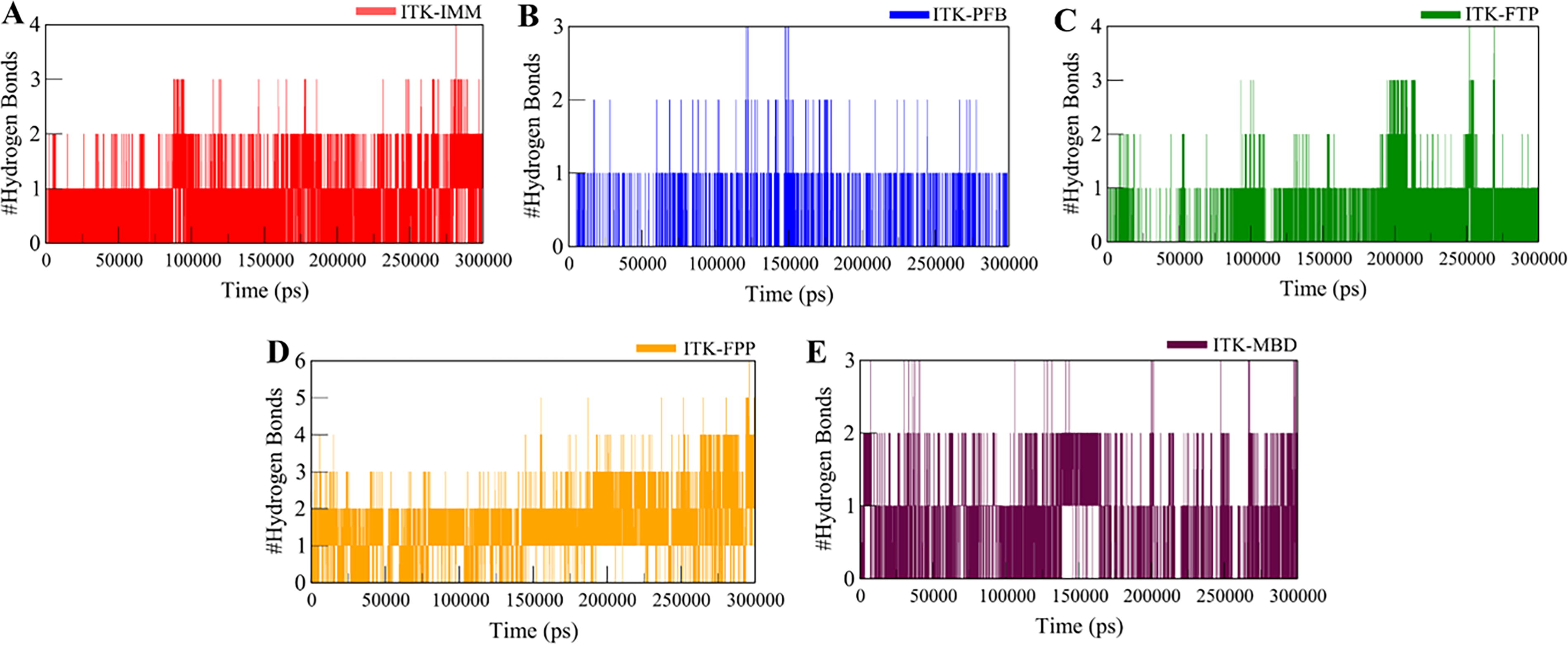
Time evaluation and stability of intermolecular hydrogen bonds formed between ITK and
Analysis of secondary structure components
Analyzing the dynamics of a protein’s secondary structure is crucial for understanding its conformational changes and folding behavior. We evaluated the secondary structure dynamics of ITK when bound to IMM, PFB, FTP, FPP, and MBD, respectively, to investigate how these interactions affect the protein’s structural conformation. Supplementary Figure S6 depicts the time evolution of secondary structure components of ITK and its complexes. However, a small decrease in α-helix content can be seen in ITK-PFB, ITK-FTP, and ITK-MBD complexes, which primarily contributed to α-helix conversion into Coils, Turn, and Bend structures. Furthermore, a minor decrease was observed in the β-sheets of ITK-FTP, suggesting less stability (Supplementary Table S6).
Principal component analysis and free energy landscapes
Principal component analysis (PCA) is a widely used statistical technique in drug discovery to study the conformational projections of protein molecules (Stein et al., 2006). Here, PCA was used to analyze the conformational projections of ITK and its complexes with IMM, PFB, FTP, FPP, and MBD. The conformational exploration of these ITK-ligand complexes was performed using projections of the Cα atoms (Fig. 4A). The results indicated that all ITK-ligand complexes exhibited slight deviations from the conformational subspace occupied by free ITK, extending beyond its principal eigenvectors. Notably, the subspaces corresponding to ITK-IMM and ITK-PFB largely overlapped with that of free ITK.
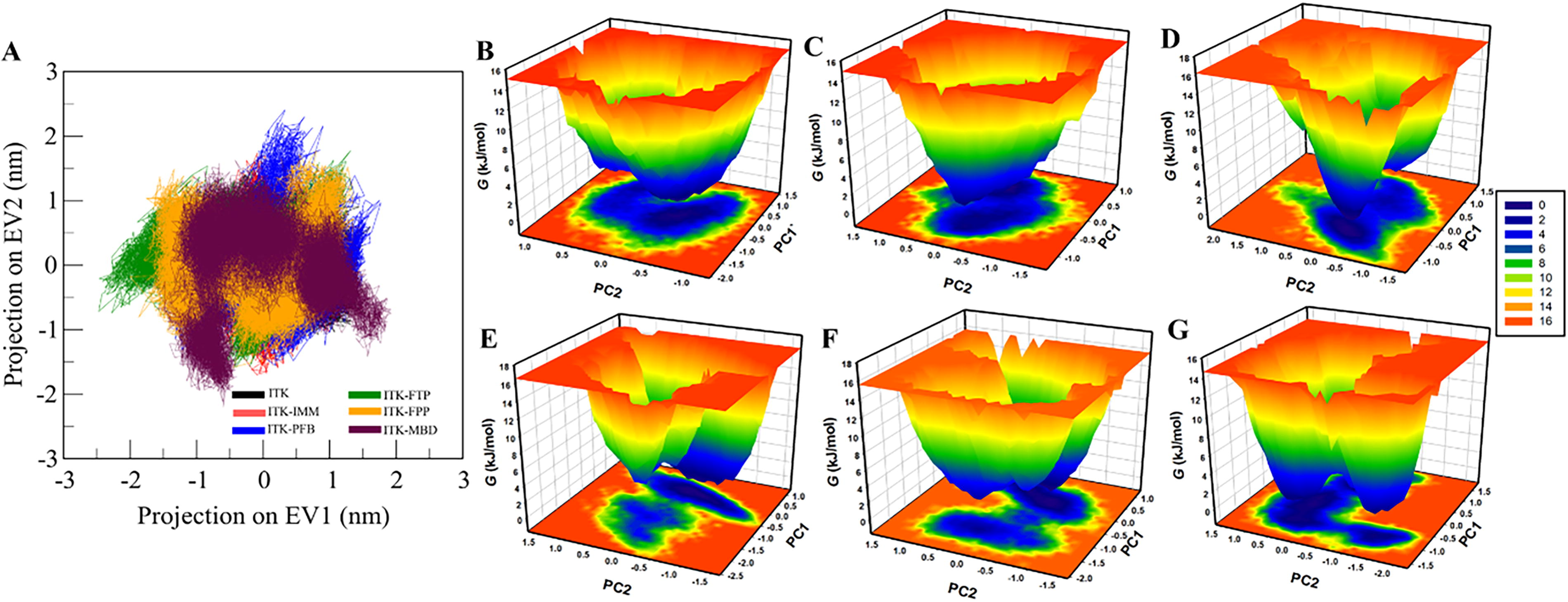
Essential dynamics.
Free energy landscape (FEL) analysis is another powerful approach used in drug discovery to understand the folding dynamics of protein molecules (Papaleo et al., 2009). The FEL plots of free ITK and its ligand-bound forms are illustrated in Figure 4B–E. In these diagrams, regions with deep blue coloration represent areas of low energy, indicating a proximity to the native state. Free ITK displayed a single, well-defined basin corresponding to its global minimum energy state. Similarly, ITK-IMM and ITK-PFB complexes also exhibited a single predominant basin, whereas the ITK-FTP and ITK-FPP complexes displayed two smaller basins. This suggests that ligand binding caused subtle shifts in the global minimum energy state of ITK.
MM-PBSA analysis
MM-PBSA is a widely used method in drug discovery for estimating the binding-free energy of protein–ligand complexes (Valdés-Tresanco et al., 2021). This thermodynamic parameter quantifies the energy change associated with complex formation and serves as an indicator of the interaction strength between a protein and its ligand. The key binding energy components and their corresponding standard deviations, are summarized in Supplementary Table S7. The findings suggest that the ITK-ligand complexes exhibit favorable binding free energies, highlighting their stability and strong intermolecular interactions. The complexes ITK-FPP and ITK-PFB had comparatively higher binding energies, indicating the formation of a more stable complex. The identified compounds PFB and FTP demonstrated binding affinities and pharmacokinetic profiles superior to existing ITK inhibitors, such as ITK-inhibitor-2. Additionally, their distinct interaction patterns, characterized by enhanced specificity within the ITK binding pocket, highlight their potential to minimize off-target effects. Compared to known ITK inhibitors, the identified compounds represent promising candidates for further therapeutic development, particularly in immune-related disorders and cancers. These findings underline the potential of computational drug design to advance next-generation ITK inhibitors.
Conclusions
The present study supports the potential of the compounds IMM, PFB, FTP, FPP, and MBD for future discovery and clinical development of drugs targeting ITK in the field of immunology. Notably, two-dimensional and three-dimensional interaction analysis confirmed stable interactions between these compounds and key residues within the ITK binding site. MD simulations provided deeper insights into the stability of ITK-ligand complexes. The RMSD, RMSF, Rg, and SASA analyses showed that the selected compounds, especially ITK-IMM and ITK-PFB, maintained structural stability and compactness throughout the 300 ns simulation period. Additionally, the hydrogen bond analysis confirmed that these interactions did not significantly disrupt the overall stability of ITK.
In all, the study showed that PFB and FTP have more optimal binding affinity and ADMET properties compared to the reference ITK inhibitors, such as ITK-inhibitor-2. Furthermore, structural analysis revealed the interaction of proposed compounds and active site residues is highly similar to known inhibitors but share distinct interaction patterns that could improve specificity. This specificity and optimization hold potential for the development of next-generation ITK inhibitors with possible applications in the treatment of immune-related disorders and cancers. Despite these advances, certain limitations must be acknowledged. The study relies solely on computational predictions, which require experimental validation through in vitro and in vivo studies to confirm biological activity and therapeutic potential. Additionally, the clinical relevance of these compounds must be further assessed, particularly concerning pharmacokinetics and potential off-target effects in complex biological systems.
Footnotes
Acknowledgments
M.I.H. acknowledges the Council of Scientific and Industrial Research for financial support [Project No. 27(0368)/20/EMR-II]. M.F.A. acknowledges and extends his appreciation to the Researchers Supporting Project Number (RSP 2025R122),
Data Availability Statement
All data generated or analyzed during this study are included in the article.
Authors’ Contributions
Conceptualization: S.A. and M.I.H. Methodology: S.A., T.M. Software: M.U.S. Validation: A.C., D.K.Y., and S.A. Formal analysis: M.F.A. Investigation: A.H. Resources: M.F.A. Data curation: A.C., T.M., A.H., and S.A. Writing—original draft preparation: S.A., M.U.S., A.C. Writing—review and editing: A.H., M.F.A., and M.I.H. Visualization: T.M., A.C. Supervision: M.I.H. Project administration: T.M., M.F.A. Funding acquisition: M.F.A. All authors made a significant intellectual contribution and have read and agreed to the final published version of the article.
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
This work is supported by Central Council for Research in Unani Medicine (CCRUM), Ministry of AYUSH, Government of India (Grant No. 3-69/2020-CCRUM/Tech). Researchers Supporting Project Number (RSP 2025R122), King Saud University, Riyadh, Saudi Arabia.
Abbreviation Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.