
Abstract
Innovation in drug discovery for human diseases stands to benefit from systems science and next-generation phenomics approaches. An example is idiopathic pulmonary fibrosis (IPF) that is a chronic pulmonary disorder leading to respiratory failure and for which preventive and therapeutic medicines are sorely needed. Matrix metalloproteinases (MMPs), particularly MMP1 and MMP7, have been associated with IPF pathogenesis and are thus relevant to IPF drug discovery. This study evaluates the comparative therapeutic potentials of doxycycline, pirfenidone, and nintedanib in relation to MMP1 and MMP7 using molecular docking, molecular dynamics simulations, and a next-generation phenomics approach. Adsorption, distribution, metabolism, excretion, and toxicity analysis revealed that doxycycline and nintedanib adhered to Lipinski’s rule of five, while pirfenidone exhibited no violations. The toxicity analysis revealed favorable safety profiles, with lethal dose 50 values of doxycycline, pirfenidone, and nintedanib being 2240kg, 580, and 500 mg/kg, respectively. Homology modeling validated the accuracy of the structures of the target proteins, that is, MMP1 and MMP7. The Protein Contacts Atlas tool, a next-generation phenomics platform that broadens the scope of phenomics research, was employed to visualize protein contacts at atomic levels, revealing interaction surfaces in MMP1 and MMP7. Docking studies revealed that nintedanib exhibited superior binding affinities with the candidate proteins (−6.9 kcal/mol for MMP1 and −7.9 kcal/mol for MMP7) compared with doxycycline and pirfenidone. Molecular dynamics simulations further demonstrated the stability of protein–ligand complexes. These findings highlight the notable potential of nintedanib in relation to future IPF therapeutics innovation. By integrating in silico and a next-generation phenomics approach, this study opens up new avenues for drug discovery and development for IPF and possibly, for precision/personalized medicines that consider the molecular signatures of therapeutic candidates for each patient.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic and progressive interstitial lung disease characterized by the irreversible scarring of lung tissue, leading to impaired lung function and ultimately respiratory failure. The exact etiology of IPF remains elusive, although a complex interplay of genetic and environmental factors, and abnormal wound healing responses are thought to play a role (Richeldi et al., 2017). Histopathological features typically include the presence of fibroblastic foci and excessive deposition of extracellular matrix (ECM) components such as collagen, resulting in lung tissue remodeling and loss of function. Despite advances in understanding its pathogenesis and the introduction of novel therapies, IPF continues to pose significant challenges owing to the paucity of therapeutic options with robust potential and safety and by extension, uncertainties in diagnosis, clinical management, and prognosis (Spagnolo et al., 2021). Early detection, drug discovery, and accurate prognostication of disease and treatment outcomes are sorely needed.
Matrix metalloproteinases (MMPs) have emerged as key players in the pathogenesis and progression of IPF. MMPs, particularly MMP1 and MMP7, are implicated in the dysregulated ECM turnover observed in IPF. MMP1, also known as interstitial collagenase, contributes to the degradation of collagen, thereby influencing tissue remodeling and fibrosis (Rosas et al., 2008). Similarly, MMP7, a matrilysin, is involved in the proteolytic cleavage of various ECM proteins, cytokines, and growth factors, modulating the inflammatory and fibrotic processes in the IPF lungs (Jaffar et al., 2022). Elevated levels of MMP1 and MMP7 have been detected in the serum and bronchoalveolar lavage fluid of IPF patients, correlating with disease severity and progression (Rosas et al., 2008; Dancer et al., 2011). Consequently, these MMPs hold veritable promise as potential molecular targets and putative molecular biomarkers for IPF diagnosis, prognostication, and therapeutics and call for further research to inform drug discovery and development.
Innovation in drug discovery for IPF and human diseases stands to benefit from systems science (Tognolini et al., 2024; Imakura et al., 2023) and next-generation phenomics approaches (Dasgupta, 2024). While there are multiple ways in which next-generation phenomics can be conceptualized, it offers a broadening in the scope of phenomics inquiries and in ways that combine structure and function, and temporal and spatial variations in phenotypes (Dasgupta, 2024). This article argues that employing one or more of these features in next-generation phenomics can help toward new strides in IPF drug discovery.
Doxycycline, a broad-spectrum MMP inhibitor, has demonstrated significant potential in preclinical models of pulmonary fibrosis by attenuating ECM deposition and fibroblast activation (Mishra et al., 2011). Pirfenidone and nintedanib, Food and Drug Administration (FDA)-approved drugs for IPF, exert their antifibrotic effects through multiple mechanisms, including inhibition of profibrotic cytokines and reducing the accumulation of ECM proteins (Finnerty et al., 2021). This study evaluated the comparative therapeutic potentials of doxycycline, pirfenidone, and nintedanib in relation to MMP1 and MMP7 using molecular docking, molecular dynamics simulations, and a next-generation phenomics approach. The overall study design is depicted in Figure 1.
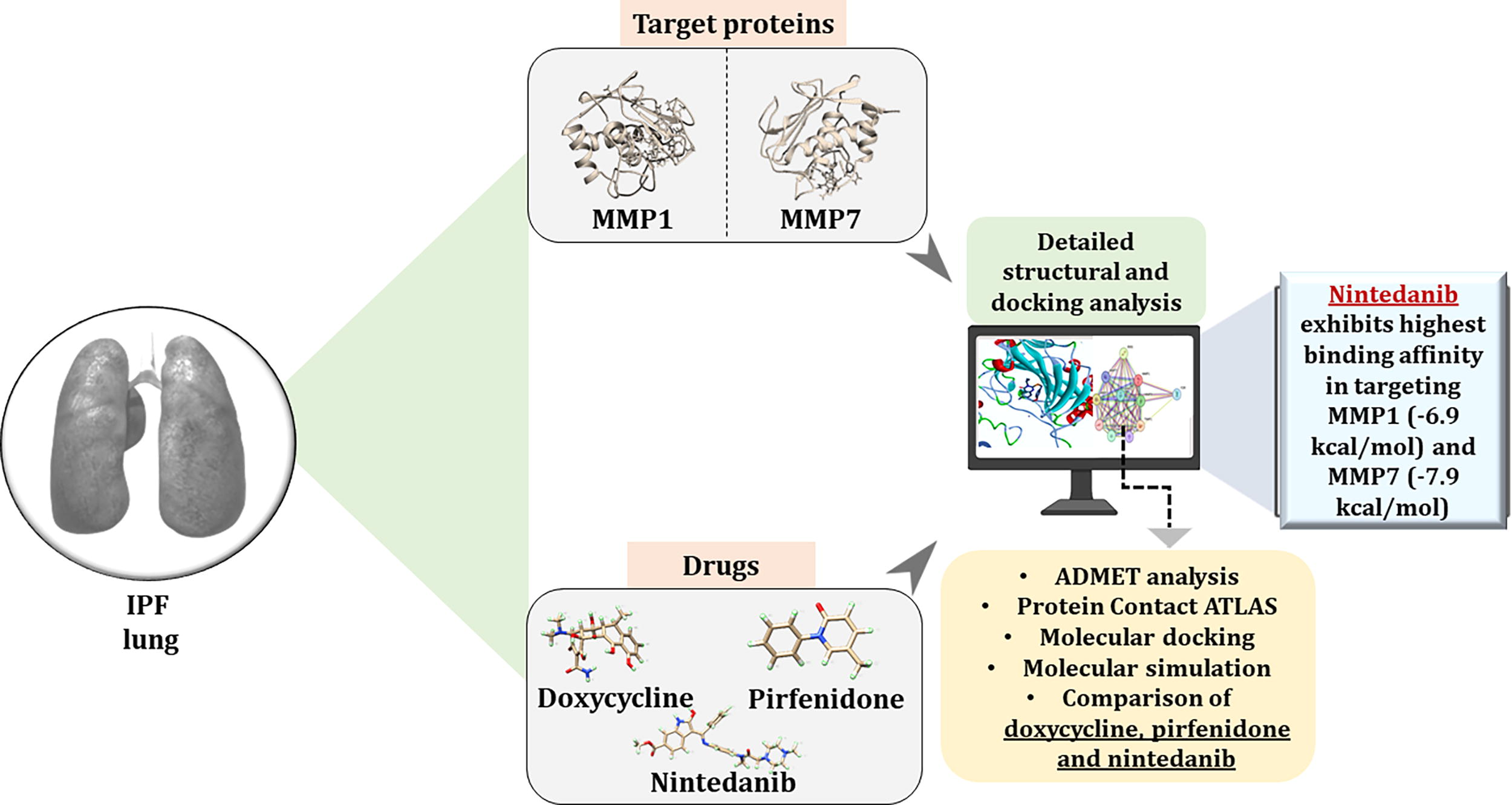
Workflow of the study design.
Materials and Methods
The present study used in silico methods and data that do not require institutional review board approval. The study was conducted under the overall research ethics oversight of the author’s institution.
Ligand selection
The selected ligands’, doxycycline, pirfenidone, and nintedanib, three-dimensional structures in simple data format were retrieved from the PubChem server (Cheng et al., 2014). The selection of these three candidate drugs was further supported by data from the DGIdb and DynaMedex databases, as well as a comprehensive review of the relevant literature (Bhattacharyya et al., 2009; Aimo et al., 2022; Ruaro et al., 2023).
Adsorption, distribution, metabolism, excretion, and toxicity studies
Adsorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis plays a pivotal role in evaluating the pharmacokinetic and pharmacodynamic properties of drug candidates, ensuring their potentials and safety profiles align with therapeutic requirements. To analyze these properties for the three selected candidate drugs, SWISSADME, a comprehensive web-based tool, was employed. This platform facilitates the assessment of a wide array of molecular descriptors and pharmacokinetic parameters, including solubility, permeability, metabolic stability, and potential toxicity. The Simplified Molecular Input Line Entry System representations of the ligands, retrieved from the PubChem database, were uploaded to SWISSADME for detailed computational analysis. The results provided critical insights into the ADMET profiles of the compounds, aiding in the identification of promising candidates for further validation (Daina et al., 2017).
Toxicity analysis
Toxicity analysis was carried out using the ProTox III web tool, a reliable platform for predicting toxicological properties based on molecular structure. This tool, accessible online, assessed various toxicity parameters, including lethal dose 50 (LD50), providing crucial insights into the safety profiles of the compounds. The analysis provided detailed insights into their potential safety risks and toxicological profiles, offering a foundation for further evaluation. These predictions play a crucial role in determining the suitability of these compounds for therapeutic applications (Banerjee et al., 2018).
Selection of target proteins
This study focused on two candidate proteins, MMP1 and MMP7, which are implicated in the pathogenesis of IPF. To support this, three transcriptomic datasets (GSE10667, GSE199152, and GSE24206) from the National Center for Biotechnology Information (NCBI)-Gene Expression Omnibus (GEO) database were analyzed to identify upregulated protein signatures associated with IPF.
GSE10667: This dataset includes 23 samples obtained from surgical remnants of biopsies or lungs explanted from patients with IPF who underwent pulmonary transplants. Among these, 8 samples were from patients with acute exacerbations of IPF, and 15 were normal lung tissues taken from disease-free margins of lung cancer resections. The morphological diagnosis of IPF was based on microscopic findings consistent with usual interstitial pneumonia (UIP) (Konishi et al., 2009; Rosas et al., 2008; Vuga et al., 2013; Yamashita et al., 2011).
GSE199152: The dataset comprises transcriptome profiling (RNA-seq) data from lung tissue biopsies of patients with IPF-UIP, rheumatoid arthritis-associated interstitial lung diseases (RA-ILD or RA-UIP), and non-UIP controls. Total RNA was extracted using the RNeasy kit (Qiagen) from fresh frozen lung tissues provided by the Lung Tissue Repository Consortium. The RNA samples were sequenced using Illumina mRNA-Seq protocols, probing a total of 23,295 genes.
GSE24206: This dataset includes whole lung samples from 11 IPF patients undergoing diagnostic surgical biopsy or transplantation, with samples taken from different lobes whenever possible. Normal controls consisted of healthy organs donated for transplantation (Meltzer et al., 2011).
The expression levels of MMP1 and MMP7 were found to be significantly upregulated in IPF patients across these datasets (Supplementary Fig. S1). These proteins were subsequently identified as key targets associated with the pathogenesis of IPF.
Homology modeling of target proteins MMP1 and MMP7
Homology modeling also plays a crucial role in generating accurate three-dimensional protein structures, which further enrich our understanding of protein–drug interactions (Pinzi and Rastelli, 2019). SWISSMODEL, an online tool, was utilized for homology modeling of the target proteins MMP7 and MMP1 (Waterhouse et al., 2018). The modeling was based on the amino acid sequences of the proteins in FASTA format:
The accuracy of the protein models generated was assessed using the PROCHECK tool (Gundampati et al., 2012). The structure is then subjected to deep optimization and validated by structure validation tools VERIFY3D, ERRAT, and QMEAN. Finally, the protein was visualized by Swiss-PDB Viewer (Hasan et al., 2015).
Protein network analysis
Protein–protein interactions (PPI) are pivotal in systems biology for evaluating target protein function and drug ability. PPI networks, comprising a heterogeneous protein network connected by edges representing interactions, with nodes denoting proteins, and adjacent nodes representing physically interacting proteins, facilitate this evaluation. STRING, a precomputed database (http://string-db.org), that is used to evaluate PPI was utilized to visualize the connected proteins of MMP1 and MMP7 (Szklarczyk et al., 2019).
Physicochemical properties and the abundance of amino acids in protein
Analysis of the physicochemical properties and amino acid composition of the target proteins, MMP1 and MMP7, was conducted using the ProtParam tool. This evaluation provided insights into the abundance of specific amino acids within their sequences, contributing to a deeper understanding of their structural and functional characteristics. These predictions are essential for assessing the stability, solubility, and potential biological activity of the proteins, thereby facilitating their characterization and elucidating their roles in disease mechanisms.
Protein Contacts Atlas
Protein Contacts Atlas, an interactive web-based database, was utilized for visualizing interactions between ligands, small molecules, proteins, and nucleic acids. This tool facilitated the visualization of molecule structures at atomic resolutions, providing valuable insights into molecular interactions (Kayikci et al., 2018). Integrating such high-resolution molecular data aligns with the principles of next-generation phenomics, which emphasize the use of advanced structural and molecular tools to uncover the intricate phenotypic consequences of genetic variations. By bridging molecular interaction data with phenotypic outcomes, this approach contributes to a deeper understanding of complex biological systems and the development of precision-targeted therapies.
Receptor/protein preparation for molecular docking
The preparation of the target proteins involved following the procedure outlined by Tallei et al. (Tallei et al., 2020). Specifically, the crystal structure of MMP7 and MMP1, serving as the target receptor, was retrieved from the Research Collaboratory for Structural Bioinformatics (RCSB) with Protein Data Bank (PDB) ID, 7WXX, and 3SHI. AutoDock Vina software was used for docking studies as per the methodology described by Trott and Olson (Trott and Olson, 2010).
Molecular dynamics simulation
Molecular dynamics simulations were conducted employing the GROMACS software suite, a widely used tool for simulating the dynamics of biomolecular systems (Vieira et al., 2023). The simulations utilized a comprehensive force field to accurately model the interactions between atoms and molecules, allowing for the investigation of the system’s behavior over time. Parameters such as temperature, pressure, and solvent conditions were carefully controlled to mimic physiological conditions. The trajectories obtained from the simulations provided valuable insights into the structural dynamics, thermodynamics, and kinetics of the system under study. Analysis of the trajectories revealed key conformational changes, protein–ligand interactions, and dynamic properties, shedding light on the underlying mechanisms governing the system’s function.
Results
ADMET studies of candidate drugs
In the present study, the pharmacokinetic and physiochemical properties of doxycycline, pirfenidone, and nintedanib were investigated. It was found that doxycycline and nintedanib follow Lipinski’s rule of one, while pirfenidone exhibited no rules of violation. It is reported that the pivotal prerequisite to determining the apparent potential of an oral drug is determined by its intestinal absorption (Huang et al., 2019). This study indicated that doxycycline has low gastrointestinal (GI) absorption, while both pirfenidone and nintedanib have high GI absorption. A bioavailability score quantifies the proportion of a drug that enters systemic circulation when administered via a specific route, typically oral ingestion. It is an important pharmacokinetic parameter to consider in drug development and clinical practice. A score closer to 1.0 indicates high bioavailability, suggesting that a significant portion of the administered drug reaches systemic circulation intact, potentially leading to therapeutic potential. The bioavailability scores of doxycycline, pirfenidone, and nintedanib are 0.11, 0.55, and 0.55, respectively (Supplementary Table S1).
Structural and toxicity analysis of drugs
The balls and sticks representation of doxycycline, pirfenidone, and nintedanib is shown in Supplementary Figures S2, S3, S4. The white, red, gold, green, and violate balls and sticks represent carbon-hydrogen bonds, hydrophobic interactions (Pi-alkyl/alkyl-interaction), hydrophobic bonds (Pi-sulfur), hydrogen bonds, and hydrophobic bonds (Pi-Pi/Pi-sigma/amide-Pi-interactions).
The toxicity analysis suggested that the LD50 value of doxycycline, pirfenidone, and nintedanib are found to be 2240, 580, and 500 mg/kg, respectively. Toxicity analysis also disclosed that the value of distribution of dose value is 2440 (doxycycline), 580 (pirfenidone), and 500 (nintedanib), respectively.
Verification and validation of the target protein structures
The RCSB (https://www.rcsb.org/) PDB was used to retrieve the X-ray crystallographic structure of MMP1 and MMP7 (Supplementary Fig. S5A and (S5B). The Ramachandran plots for glycine, proline, and pre-proline residues are shown in Figure 2A–C. The QMEAN Z-score serves as a measure of how closely the structural features in a model resemble those found in experimental structures, providing an assessment of its “nativeness” on a broad scale. It gauges whether the model’s QMEAN score aligns with what would be expected for similar-sized experimental structures. A QMEAN Z-score near zero indicates high quality, indicating that the model’s structure closely resembles the experimental ones. Conversely, scores of −4.0 or lower suggest poor-quality models. The QMEAN scores for MMP1 and MMP7 were 0.73 and 0.77, respectively (Fig. 2D). The quality factor predicted by the ERRAT server for both MMP1 and MMP7 are 72.87 and 77.46. VERIFY3D server predicted that 92.68% (MMP1) and 98.93% (MMP7) of the residues have averaged 3D-1D score ≥0.1.
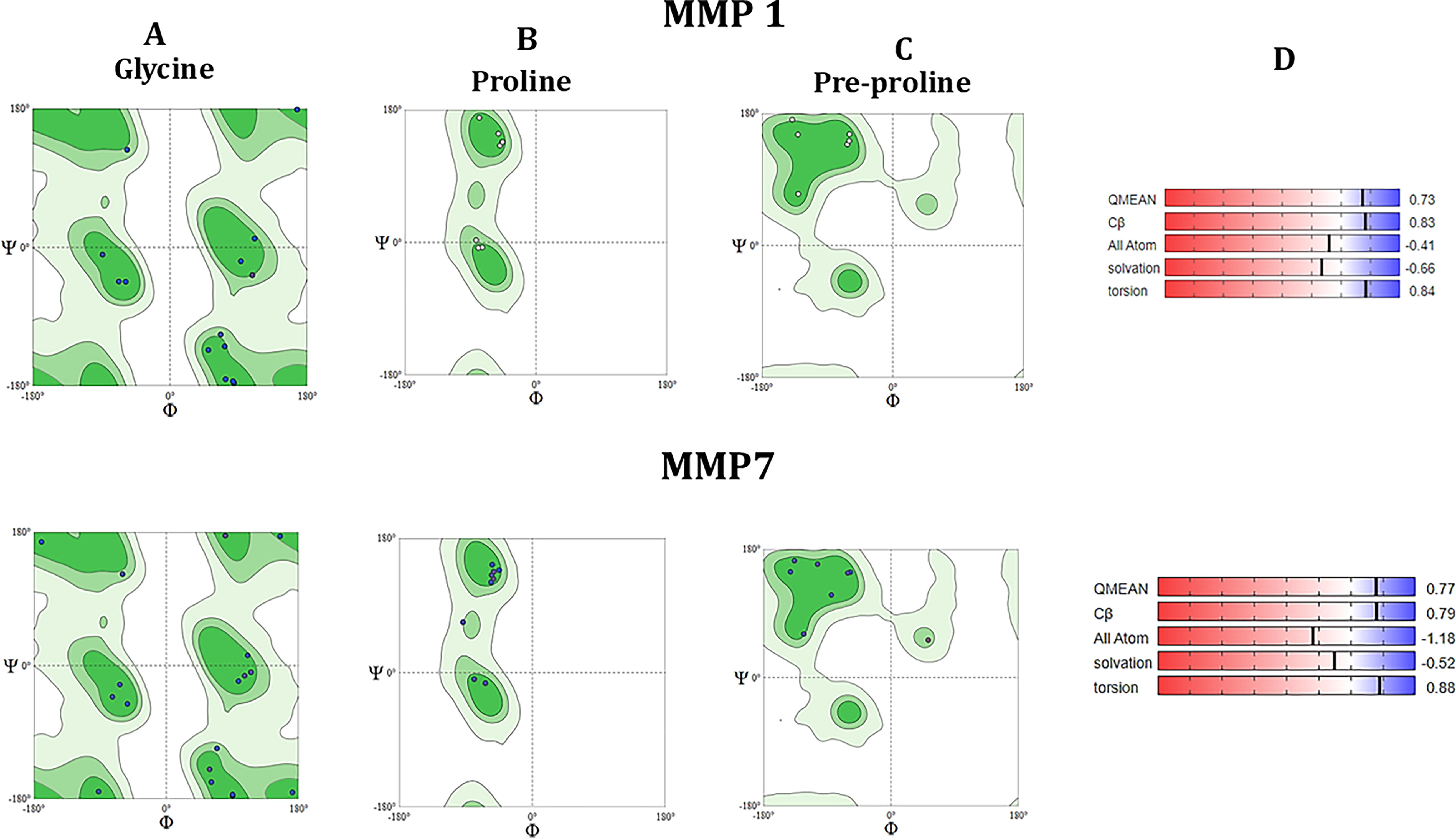
Ramachandran plot. Ramachandran plot [
The three-dimensional geometry of the protein model was further validated by the PROCHECK web tool, which calculated the Ramachandran plot and generated results for residues showing regions with different colors, that is, red (favored), yellow (additionally allowed), pale yellow (generously allowed), and white colored areas (disallowed) (Supplementary Fig. S6). The plot for MMP1 demonstrated that 45 glycine and 27 proline residues exist. In the case of MMP7, the plot shows a total of 20 glycine and 9 proline residues are present. The R-factor, which predicts the quality of the protein model, is calculated. The R-factor of 0.215 for MMP1 and 0.17 for MMP7 suggests that the models are of good quality as an R-factor value < 20.0 is typically associated with a high-quality model, which is expected to have over 90% of residues in the most favored regions.
Protein Contacts Atlas
In this study, at the atomic levels, the protein contact visualization was studied using the Protein Contacts Atlas tool. Protein Contacts Atlas is a cutting-edge tool that facilitates atomic-level visualization and analysis of protein interactions, leveraging high-resolution structural data from repositories like the PDB. This tool exemplifies next-generation phenomics by providing precise mapping of interatomic contacts, including hydrogen bonds, hydrophobic interactions, and salt bridges, enabling detailed insights into protein function and dynamics. It emphasizes the structural determinants of protein behavior, bridging the gap between molecular structure and phenotype while advancing traditional phenomics approaches.
Protein Contacts Atlas chord plots for MMP1 and MMP7 provide a detailed visualization of protein contacts at the secondary structure level (Fig. 3A). Similarly, the atomic neighborhood of the selected residues or ligands is represented by an asteroid plot (ligand/residue-centric view). The asteroid plot and scatter plots of both proteins are shown in Figure 3B and C, respectively. Both of these plots provide an insight into the quantitative characteristics per residue.

Visualization of protein contacts and atomic neighborhood for MMP1 and MMP7.
Physicochemical properties and amino acid composition of MMP1 and MMP7
MMP1 (UniProt ID: P03956) consists of 469 amino acids with a molecular weight of 54,006.90, a theoretical pI of 6.47, and an atomic composition of C: 2458, H: 3666, N: 656, O: 699, and S: 13. The amino acid composition includes Gly (7.5%), Phe (7.7%), and Asp (7.0%) as the most abundant residues, with Cys (0.6%) being the least. It has 58 negatively charged residues (Asp + Glu) and 54 positively charged residues (Arg + Lys). MMP7 (UniProt ID: P09237) comprises 267 amino acids with a molecular weight of 29,676.84, a theoretical pI of 7.73, and an atomic composition of C: 1330, H: 2053, N: 361, O: 387, and S: 12. Its amino acid composition highlights Leu (10.1%) and Gly (9.7%) as the most abundant residues, while Cys (1.1%) is the least. The protein includes 28 negatively charged residues (Asp + Glu) and 29 positively charged residues (Arg + Lys).
Protein interaction network
In the present study, the protein network of MMP1 and MMP7 was studied using the STRING database. The specific lines of evidence are represented by line colors linking the protein nodes that are involved in creating functional association whereas the confidence is denoted by the distance between nodes as recognized by the Bayesian scoring system (Supplementary Fig. S7). The PPI enrichment p-value of MMP1 and MMP7 were 8.92e-11 and 1.64e-12, respectively.
Docking of drugs into MMP1 and MMP7
Docking analysis is mainly performed to determine the interaction between the MMP1 and MMP7 protein molecules and three ligands (doxycycline, pirfenidone, and nintedanib). The molecular docking analysis between doxycycline, pirfenidone and nintedanib, and MMP1 exhibited a binding affinity score of −6.1, −6.6, and −6.9 kcal/mol, respectively. In the case of MMP7 the binding affinity score was −6.4 kcal/mol (doxycycline), −6.8 kcal/mol (pirfenidone), and −7.9 kcal/mol (nintedanib). The detailed docked structure of MMP1 and MMP7 proteins with the three candidate drugs are shown in Figures 4 and 5.
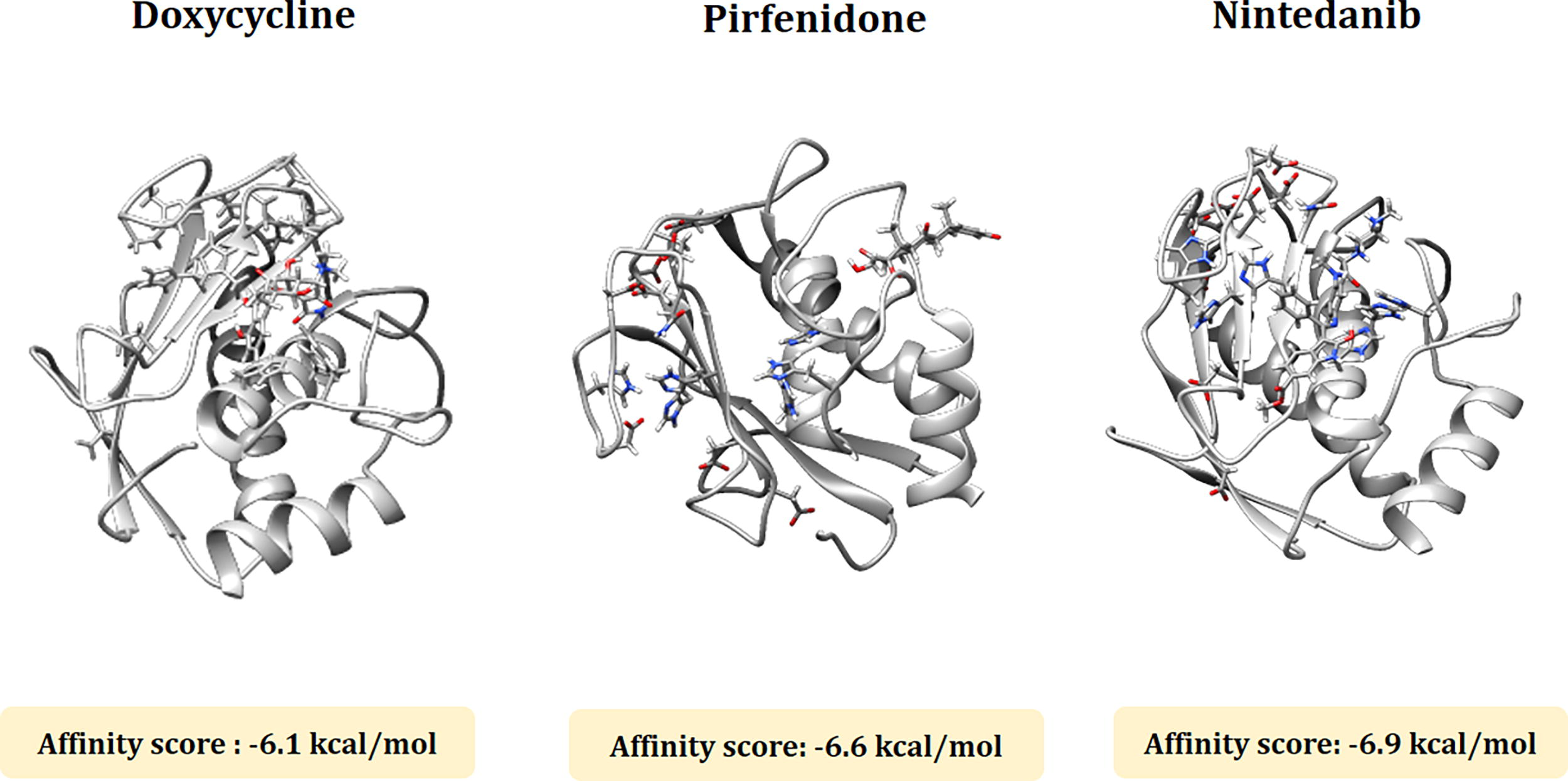
The molecular docking analysis between doxycycline, pirfenidone, and nintedanib and MMP1. The binding affinity scores of doxycycline, pirfenidone, and nintedanib with MMP1 are −6.1, −6.6, and −6.9 kcal/mol respectively.
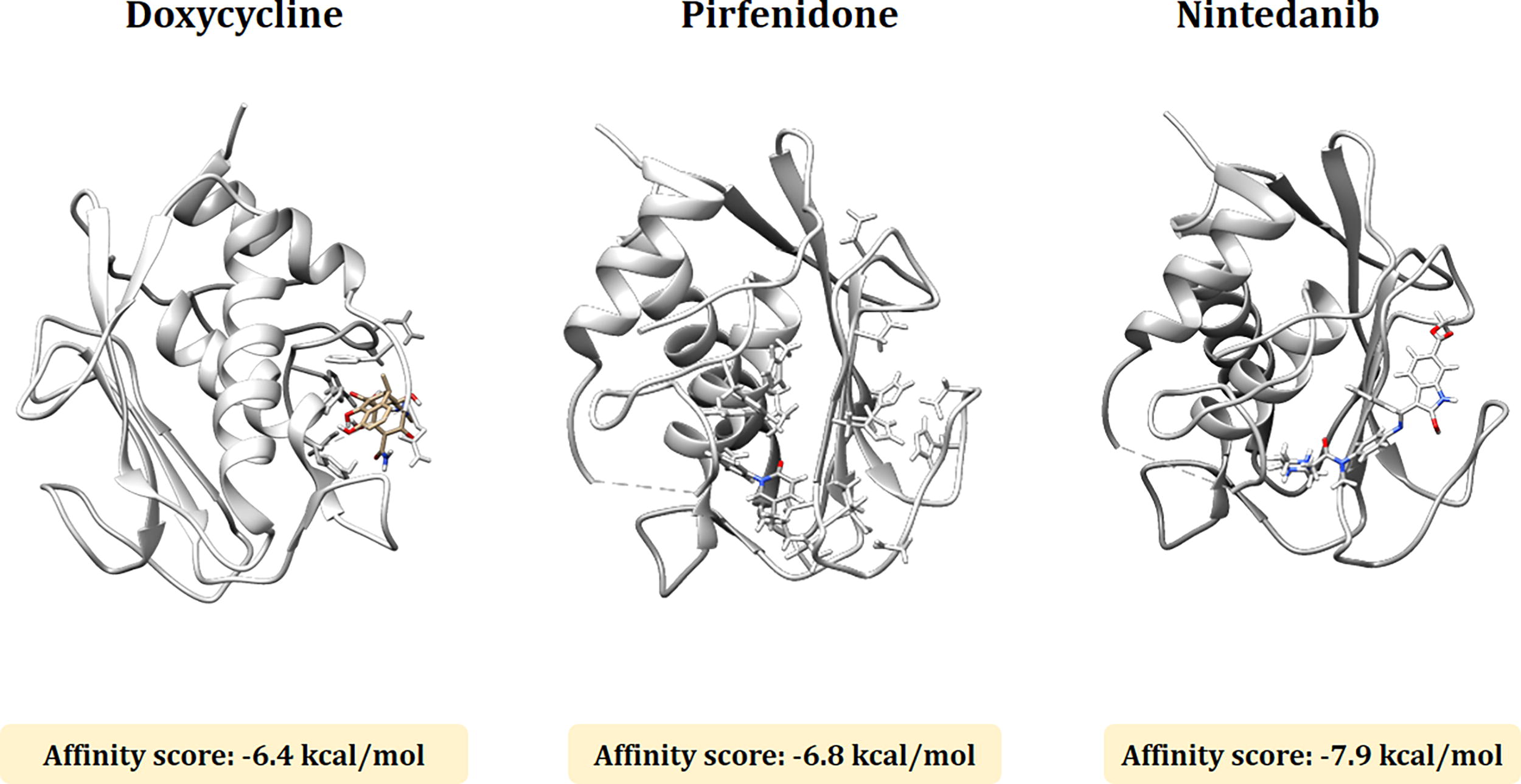
The molecular docking analysis between doxycycline, pirfenidone, and nintedanib and MMP7. The binding affinity score with MMP7 is −6.4 kcal/mol (doxycycline), −6.8 kcal/mol (pirfenidone), and −7.9 kcal/mol (nintedanib).
Molecular dynamic simulation of docked structure
In this study, molecular dynamic simulations of the docked structures were performed to assess structural stability. The radius of gyration (Rg) and root mean square deviation (RMSD) values were used to evaluate any deviation in the docked proteins’ conformation over a 400-ps simulation. The Rg that measures protein compactness was found to be 1.50 nm for MMP1 (400 ps) and 1.55 nm for MMP7 (400 ps). The trajectory RMSD values were found to be 0.0004–1.079 nm for MMP1 (400 ps) and 0.0004–6.92 nm for MMP7 (Supplementary Fig. S8).
Discussion
IPF is in need of new drugs and treatment options. The most salient finding of the present study is the notable potential of nintedanib in relation to future IPF therapeutics innovation. In silico research, systems science coupled with phenomics offer new possibilities for IPF drug discovery. Historically, it is interesting to note that phenomics innovation has lagged behind other omics and multi-omics technologies. Addressing this “phenomics gap” is crucial for making strides toward a better understanding of the pathogenesis of complex diseases such as IPF. Next-generation phenomics, as noted in the introduction, offers a transformative approach by integrating high-throughput technologies and computational tools to analyze phenotypic traits at unprecedented spatial and temporal resolution (Dasgupta, 2024). The present integrated approach of in silico research, next-generation phenomics, and systems biology can provide valuable insights into IPF management and accelerate the development of targeted, personalized therapies.
This study compared the potential of three promising drugs, that is, doxycycline, pirfenidone, and nintedanib, in targeting two important key players of IPF (MMP1 and MMP7) using next-generation phenomics coupled with molecular docking analysis. The structural integrity of MMP1 and MMP7 proteins, key targets in IPF, was validated through a series of quality assessments using tools such as QMEAN Z-scores, ERRAT, and VERIFY3D. The Ramachandran plots confirmed that the majority of residues in both MMP1 and MMP7 are in favored regions, further validating the reliability of the models for subsequent docking studies. The Protein Contacts Atlas tool’s chord, asteroid, and scatter plots for MMP1 and MMP7 provided a detailed visualization of non-covalent interactions and residue characteristics, offering insights into how drugs like doxycycline, pirfenidone, and nintedanib may impact protein dynamics in IPF. In the chord plot, the size of the arc is directly proportional to the number of residues in the secondary structure, while its thickness reflects the number of contacts. The circular arrangement illustrates non-covalent interactions with residues in the first shell. Notably, asteroid plots provide insights into structure-based drug design and protein engineering, emphasizing the impact of mutations. Residues in the second shell are depicted by larger concentric circles, representing neighboring residues without direct ligand interaction. In addition, scatter plots elucidate the relationship between two variables for each chain residue, indicating residue properties such as network centrality, residue extent, and complex solvent-accessible area. Residues in outlier areas of scatter plots may hold significance for protein structure, stability, and function. Integrating this high-resolution interaction data with the framework of next-generation phenomics enhances our understanding of molecular mechanisms by linking protein interaction patterns to phenotypic outcomes. Furthermore, the significant PPI enrichment value observed in the PPI network of MMP1 and MMP7 underscores a notable degree of connectivity and interplay between these MMPs.
Molecular docking, a powerful tool for assessing the binding affinity between target proteins and potential therapeutic drugs, was used in the present study to highlight the potential of the candidate drugs and targeted proteins (Ali et al., 2023). While docking studies of these drugs with various MMPs have been previously conducted, this study presents a novel analysis by focusing on the MMPs specifically involved in the pathogenesis of IPF (Dasgupta, 2024; Patnaik et al., 2021). The docking results showed that nintedanib demonstrated the strongest interactions with the two key proteins of IPF, MMP7, and MMP1, compared with pirfenidone and doxycycline. Molecular dynamics simulations further elucidated the underlying molecular mechanisms, with Rg analysis indicating stable protein structures, which suggest effective ligand binding and enhanced stability (Ghahremanian et al., 2022). In addition, the small RMSD values observed during simulations reflected minimal structural deviations, reinforcing the stability of ligand–protein interactions.
Nintedanib’s superior potential in targeting MMP7 and MMP1 aligns with its known mechanism of action as a tyrosine kinase inhibitor, which interferes with pathways involved in fibroblast activation and ECM deposition. By inhibiting tyrosine kinases such as receptors for platelet-derived growth factor, fibroblast growth factor, and vascular endothelial growth factor, nintedanib demonstrates potent antifibrotic effects (Rangarajan et al., 2016). In contrast, doxycycline, a broad-spectrum antibiotic with anti-inflammatory and MMP inhibitory properties (Palasuk et al., 2018), and pirfenidone, an antifibrotic agent modulating multiple profibrotic pathways, showed comparatively weaker effects on MMP7 and MMP1 expression levels (Chuliá-Peris et al., 2022). These findings suggest that while doxycycline and pirfenidone may attenuate fibrotic processes in IPF, their mechanisms of action may not specifically target MMP7 and MMP1 to the extent observed with nintedanib. This highlights nintedanib’s potential as a preferred treatment option for IPF patients, particularly those with elevated MMP7 and MMP1 levels.
While this study highlights the potential of nintedanib in targeting MMP7 and MMP1, a key limitation is the reliance on in silico analyses, including molecular docking and dynamic simulations, which, despite providing valuable insights, may not fully capture the complexity of in vivo biological systems. The therapeutic effects of these drugs on MMP7 and MMP1 expression need to be validated through experimental studies, such as cell-based assays and animal models, to confirm their clinical relevance. In addition, the impact of drug interactions and off-target effects, which could influence treatment outcomes, was not explored in this study. Future investigations should integrate experimental validation with clinical data to strengthen the translational potential of these findings. Another limitation of this study is the focus solely on MMP1 and MMP7 as molecular targets, while other MMPs implicated in IPF pathogenesis were not explored (Zheng et al., 2024). Future research should expand the scope by investigating additional MMPs and their role in IPF progression. Moreover, it is important to assess the therapeutic potential of other drug candidates that target these established molecular targets, potentially improving treatment outcomes and advancing personalized therapies for IPF.
Overall, the present study provides a novel approach to IPF treatment by exploring the differential effects of doxycycline, pirfenidone, and nintedanib on key proteins such as MMP1 and MMP7. While doxycycline, pirfenidone, and nintedanib are already established in clinical practice for managing IPF, this study offers new insights into their differential therapeutic potential based on specific molecular targets. The findings suggest that nintedanib may be more effective in IPF cases where MMP1 and MMP7 are significantly upregulated, offering a potential advantage over doxycycline and pirfenidone. By incorporating molecular profiling, this research highlights the potential for personalized treatment strategies in IPF, paving the way for the development of more targeted and effective therapies.
Conclusions
The key finding of this study highlights the significant potential of nintedanib in driving future innovations in IPF therapeutics, demonstrating the most prominent targeting of MMP7 and MMP1, crucial molecular markers in IPF pathogenesis, compared with doxycycline and pirfenidone. This supports its established role as a tyrosine kinase inhibitor influencing fibrotic pathways. By integrating molecular docking with next-generation phenomics tools, this study paves the way for advancing drug discovery and development for IPF. It also highlights the broader potential of precision and personalized medicine strategies, targeting candidate proteins involved in disease pathogenesis and developing therapeutic candidates tailored to individual patients.
Footnotes
Acknowledgments
The author is thankful for the permission to use the freely available figure-making tool BioRender.com and NCBI-GEO tool for the gene expression data.
Author’s Contributions
S.D.: Conceptualization, formal analysis, investigation, visualization, and writing–review/editing.
Author Disclosure Statement
The author declares she has no conflicting financial interests.
Funding Information
No funding was received for the present study.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.