
Abstract
Huntington’s disease (HD) is characterized by progressive motor dysfunction and cognitive decline. Early diagnosis and new therapeutic targets are essential for effective interventions. We performed integrative analyses of mRNA profiles from three microarrays and one RNA-seq dataset from the Gene Expression Omnibus database. The datasets included were GSE8762, GSE24250, GSE45516, and GSE64810. Data pre-processing included background correction, normalization, log2 transformation, probe-to-gene symbol mapping, and differential expression analysis. We identified 80 differentially expressed genes (DEGs) based on a significance threshold (p < 0.05) and absolute log fold change (logFC) >0.65. Additionally, we conducted Gene Ontology (GO) and pathway analyses of the identified genes. Protein-protein interactions among DEGs revealed a network from which seven hub genes (VIM, COL1A1, COL3A1, COL1A2, DCN, CXCR2, and S100A9) were identified using the cytoHubba plugin in Cytoscape software. Two top DEGs, IGHG1 (up-regulated) and PITX1 (up-regulated), also hold potential as therapeutic targets. Insofar as biological contextualization of the findings is concerned, the top enriched GO terms were skeletal system development, blood vessel development, and vasculature development. Molecular function terms highlighted signaling receptor binding, extracellular matrix structural constituent, and platelet-derived growth factor binding. Notably, the significant KEGG pathways included amoebiasis, the AGE-RAGE signaling pathway in diabetic complications, and the relaxin signaling pathway. In conclusion, the present computational biology integrative analyses of multiple datasets discovered new DEGs and seven hub genes, shedding light on molecular mechanisms of HD. These findings call for translational clinical omics research and may potentially lead to future precision medicine interventions and novel diagnostic biomarkers and therapeutic targets.
Introduction
Huntington’s disease (HD) is a highly progressive neurodegenerative disorder with a profound and destructive impact on patients and their families (Ravi, 2024). An autosomal dominant condition, HD is characterized by a triad of clinical manifestations, including motor dysfunction, cognitive decline, and psychiatric disturbances, ultimately culminating in a profound loss of independence and functionality (Haider et al., 2017). Despite significant advancements in our understanding of the genetic underpinnings of HD, no cure or disease-modifying therapies currently exist (Sampaio, 2024). Thus, there is an urgent need for innovative approaches to expedite the diagnosis, therapeutics, and clinical management of this debilitating condition.
Understanding the molecular complexities of HD is essential for developing effective therapeutic interventions. An expanded and unstable CAG causes HD repeat within the HTT gene, producing a mutant huntingtin protein (mHTT) with an elongated polyglutamine tract (Tabrizi et al., 2019). mHTT triggers a cascade of pathological events, including protein aggregation, mitochondrial dysfunction, impaired axonal transport, and transcriptional dysregulation (Jurcau, 2022). These complex molecular changes trigger neuronal dysfunction and eventual cell death, particularly in the striatum and cortex (Velusamy et al., 2017). Early diagnosis of HD is critical for improving therapeutic outcomes, as timely intervention may slow disease progression and enhance patient management. Integrating molecular biomarkers into early diagnostic frameworks could improve precision medicine strategies for HD.
In this context, over the past two decades, high-throughput genomics and multi-omics technologies have emerged as powerful tools for unraveling the molecular underpinnings of complex brain diseases (Kumar et al., 2024; Verma et al., 2024), including HD (Christodoulou and Papanicolaou, 2024). One approach that holds great promise for deeper understanding and targeted therapies is the comprehensive analysis of molecular alterations in affected tissues, particularly in the brain (Ahamad and Bhat, 2022). The arrival of high-throughput genomic technologies, such as microarrays and RNA sequencing (RNA-seq), has revolutionized the understanding of the complex genetic landscape of human diseases and offers new prospects in research concerning HD (Malla et al., 2021). The present study reports on genetic/genomic perturbations associated with HD through an integrative analysis of mRNA profiles from three microarrays and one RNA-seq dataset from the Gene Expression Omnibus (GEO) database. Furthermore, the study unveiled the hub genes of potential relevance to HD pathogenesis.
Materials and Methods
The present study is in silico research, did not involve human or animal research, and did not require informed consent and research ethics board approval. It was conducted under the overall research ethics oversight of the authors’ institutions.
Dataset and data pre-processing
We adopted certain inclusion and exclusion criteria to select the gene expression studies on HD for use in this integrated analysis. These were: (1) mRNA expression using microarray or RNA-seq, (2) provided unaltered data for further analysis, (3) had control samples in differentially expressed genes analysis, and (4) used human samples from blood, fibroblasts, or brain. There were some criteria for exclusion of studies, including: (1) studies available as abstracts, (2) if the study was based on model organisms, (3) if the study was based only on ncRNA, and (4) below cutoff value of cases-control were provided in the studies. A search for GEO datasets was done using ‘Huntington’s disease’ and ‘Huntingtin’ keywords for Homo sapiens with the limitation towards datasets that contain raw or processed expression data. The study analyses were done using R statistical software with a logical working plan (Clough and Barrett, 2016). We utilized the R statistical software for data acquisition and initial pre-processing. Specifically, we retrieved datasets with accession numbers GSE8762, GSE24250, GSE45516, and GSE64810 from the National Center for Biotechnology Information (NCBI) GEO database through a comprehensive search (Clough and Barrett, 2016). Subsequently, we downloaded the required dataset from the GEO public repository using the GEQquery R package (Davis and Meltzer, 2007).
After the update of GEO2R in November 2020, it can be first defined test group and next can be defined control group, so the base of the log fold change (logFC) follows the convention of the upregulated genes with positive values and downregulated genes with negative values. This was done under the metadata fields of Accession No., Title, and Source Name so that the sample assignment could be made repeatable. Before applying differential analysis, we conducted multiple operations with the data pre-processing. These operations include background correction, Trimmed Mean of M-values (TMM) normalization, transforming the expression profile obtained from GEO to base 2, converting the probe ID, and omitting the outlier samples from each expression profile. Another approach to improve the result’s readability was converting probe IDs to gene symbols using annotation packages provided on Bioconductor (Gentleman et al., 2004). It is found that multiple probes often correspond to the same gene symbol. Our analysis treated these probes as independent observations, utilizing standardized measures such as p values to summarize their contributions. We selected the gene symbols with the smallest p value among the probes corresponding to each symbol for further analysis. Any unmapped probe gene IDs were subsequently excluded from the count matrix.
Statistical analysis of microarray data
The analysis of differential gene expression was carried out using the limma (Ritchie et al., 2015) and edgeR (Robinson et al., 2010) packages within the R statistical software (Chambers, 2008). These packages are essential for proper differential expression tests in analyses utilizing gene expressions from the selected datasets. The limma package has the features that enable one to determine differentially expressed genes in microarray data analyses (Ritchie et al., 2015). This still involves modeling a linear value in the expression data of each gene of interest to provide the highest p value. Limma requires the design matrix to have some values with reference to the grouping variable to perform this process. Therefore, the study’s main aim is to determine the difference in mean expression between control and experimental samples for each gene. To do this, limma first computes the mean expression levels with the help of the lmFit() function that fits linear models specified by the design matrix to the data. This computation yields the mean expression levels for both control and experimental samples. Subsequently, it becomes necessary to specify the groups for comparison within limma. To accomplish this, a contrast matrix is defined using the makeContrasts() method.
The comparison of gene expression differences between experimental and control samples is assessed through the moderated t-test using the eBayes() method within the limma package in R statistical software. Finally, to control for false discoveries, the False Discovery Rate (FDR) approach (Storey, 2011), specifically employing the Benjamini–Hochberg (BH) method (Ferreira and Zwinderman, 2006), in the analysis is used to adjust each obtained p value. It assists in reducing false positive findings and allows for an increase in the confidence in the identification of statistically significant differential gene expressions. Therefore, by using the limma package, moderated t-test analysis, and FDR correction using the BH method, more accurate arrangements, methods, and practices are used to identify DEGs in the analyzed datasets.
Integrative analyses of microarray datasets
The next process includes a comparison of the p values for genes that are consistent in all the datasets to determine a list of DEGs. The above amalgamation is done using Fisher’s combined probability test method in the R Statistical tool. This method includes adding the logarithmically transformed, corrected p values for a given gene across all the datasets. The resultant sum is then compared to a
Gene ontology and pathway enrichment analysis
It is necessary to analyze the biological function of the identified DEGs when the integrative analyses are accomplished. To achieve this goal, we included and exhaustively analyzed the two cardinal aspects, namely pathway and functional enrichment. More precisely, to illustrate GO and pathway enrichment analysis, we used the gprofiler2 R package that provides enriched analysis for KEGG, REACTOME, and WikiPathways. Our study employed a criterion of FDR-corrected p value <0.05 as a way of establishing that the enriched pathways and functions derived from DEGs are statistically significant and reflective of the study.
PPI network construction and hub gene analysis
We obtained all human Protein-protein interaction (PPIs) from the STRING database (https://string-db.org/) with the help of the String app in Cytoscape v3.7.1 (Doncheva et al., 2019). To further analyze the crucial genes in the PPI network, we utilized the cytoHubba application involving no settings put into the Cytoscape software. Degree, maximum neighborhood component (MNC), and maximum clique centrality (MCC) were used along with the centrality measures like Betweenness, BottleNeck Stress and Closeness. To achieve this, these cytoHubba plug-ins in Cytoscape were used to build networks of the top 15 nodes associating the genes and facilitate the recognition of the most influential genes within the network.
Results
Dataset and exploratory analysis
The raw data from four selected datasets, each identified by the GEO accession numbers GSE8762, GSE24250, GSE45516, and GSE64810, were acquired using the GEOquery R package (Davis and Meltzer, 2007). Subsequently, these datasets underwent thorough preparation for statistical integrative analysis. The Robust Multi-Array Average (RMA) pre-processing method was applied to each microarray dataset to ensure data quality and consistency. This process included background correction and transforming expression values to a logarithmic scale (log2 change). Furthermore, to minimize variation across datasets and allow for meaningful comparisons, the expression values were subjected to TMM normalization. This process makes it easier to compare the datasets since the variables’ scales are made similar to each other during the process referred to as normalization. The flowchart for allocation of children in this study is described in Supplementary Figure S1. Box plot data was generated to confirm the efficiency of these preprocessing steps and the quality of the data (Supplementary Fig. S2). We used the boxplot to compare and assess whether the expression patterns were irregular or not. Such irregularities would indicate the necessity for further normalization before proceeding with downstream analyses.
Integrative analysis and DEGs screening
Primarily, a total of 80 DEGs were identified in all three sets (Fig. 1). The list of the identified DEGs is provided in Supplementary Table S1, where genes are arranged in ascending order of their BH-corrected p values. To get a clearer overview of the expression patterns of these DEGs, a heatmap was generated with the aid of R, as shown in Figure 1A. This heatmap displays sample patterns of the DEGs’ relative expression and provides a clear visualization of the data set. These DEGs were identified using a cut-off of greater than 1.5 FC from a randomly expected count with a p value <0.05 to indicate significance. These criteria are similar to the earlier transcriptomic research work done on neurodegenerative disorders, which will help in the proper identification of genes. As for the DEGs, re-evaluating the two criteria indicated that 20 of them met the criteria set at a BH-corrected p value <0.05 and a logFC >0.65. However, at this level of statistical significance, there were only 3 downregulated DEGs. These results are visually described and illustrated in a volcano plot depicted in Figure 1B. These results put emphasis on these DEGs and their plausible function in the investigated biological context.

Differentially expressed genes (DEGs).
Pathway and functional enrichment analysis
To get a better understanding of the biological function of the DEGs and their potential relevance to HD pathogenesis, we performed a comprehensive bioinformatics analysis. We used the GO, KEGG pathway, WikiPathway, and Reactome Pathway databases for this analysis. All these analyses were performed using the gprofiler2 R package, which is available on the Bioconductor platform. In the biological processes category, the most significantly enriched GO terms were “skeletal system development,” “blood vessel development,” and “vasculature development.” These terms suggest that the DEGs play a role in the developmental processes of skeletons and vessels. Among the molecular functions, the GO terms were “signaling receptor binding,” “extracellular matrix structural component,” and “platelet-derived growth factor binding,” indicating that these DEGs are involved in signaling and structural roles in the extracellular matrix. When it comes to cellular components, the most significantly enriched GO terms were collagen-containing extracellular matrix (GO:0060152), extracellular region (GO:0071944), and extracellular matrix (GO:0031012), which pointed to the location where these DEGs are functioning.
In terms of KEGG analysis, the significant pathways that are related to the identified DEGs include “Amoebiasis,” “AGE-RAGE signaling pathway”, and “Relaxin signaling pathway”, and these are likely to give an understanding of the possible pathways affected by these changes in gene expression. Additionally, the more specific Reactome and WikiPathways terms were “Diseases related to glycosaminoglycan metabolism,” “Scavenging by Class A Receptors,” “ECM proteoglycans,” “Burn wound healing,” “Inflammatory response pathway,” and “Type I collagen synthesis in osteogenesis imperfecta.” These findings consequently help to enrich the knowledge of the different biological processes and pathways associated with the DEGs. The detailed information regarding the enriched GO terms, KEGG pathways, and Reactome/WikiPathways is presented in Figure 2 and Figure 3. These analyses all together help in understanding the possible biological significance of the identified DEGs in relation to HD pathogenesis.
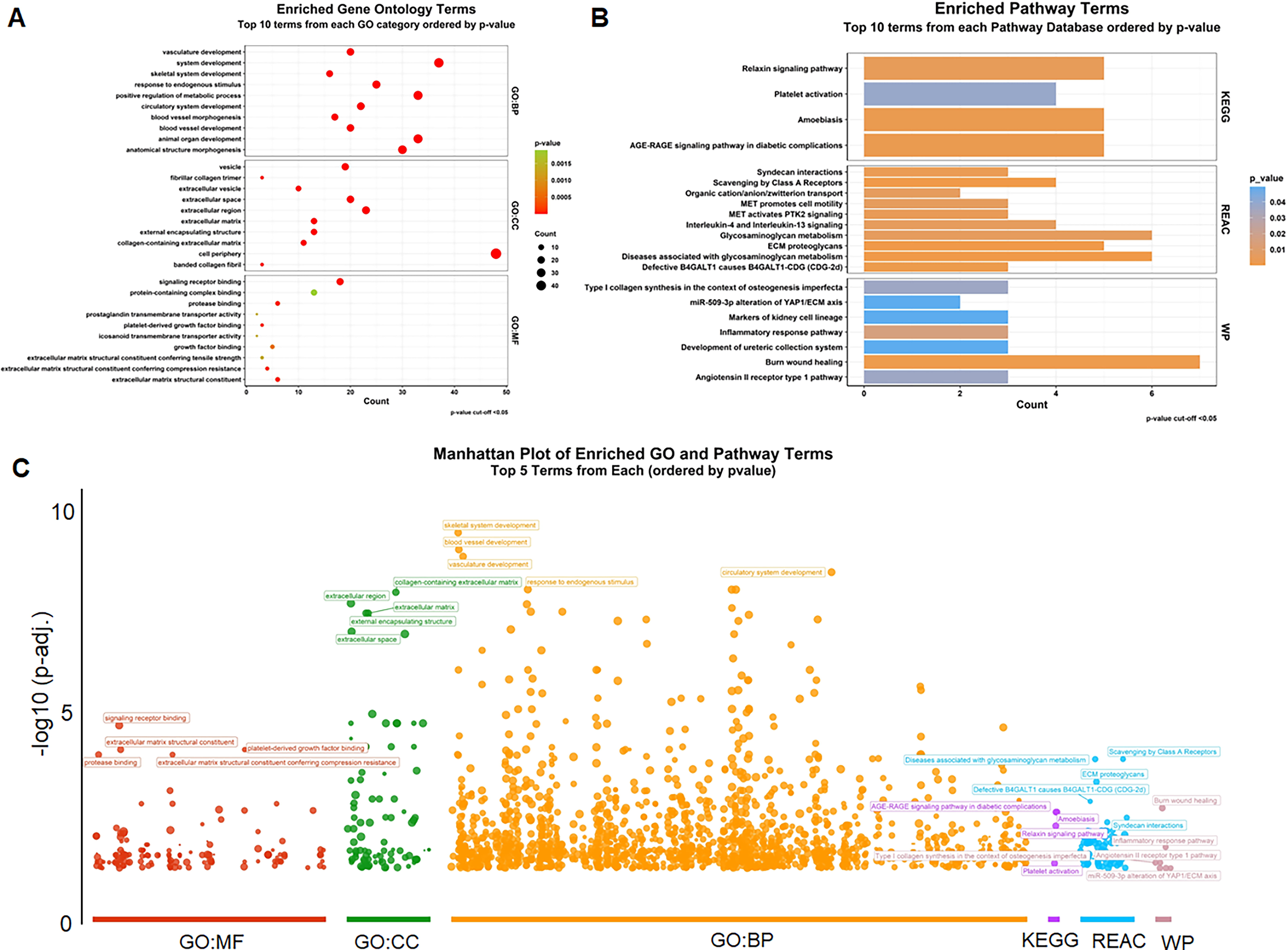
Gene enrichment analysis
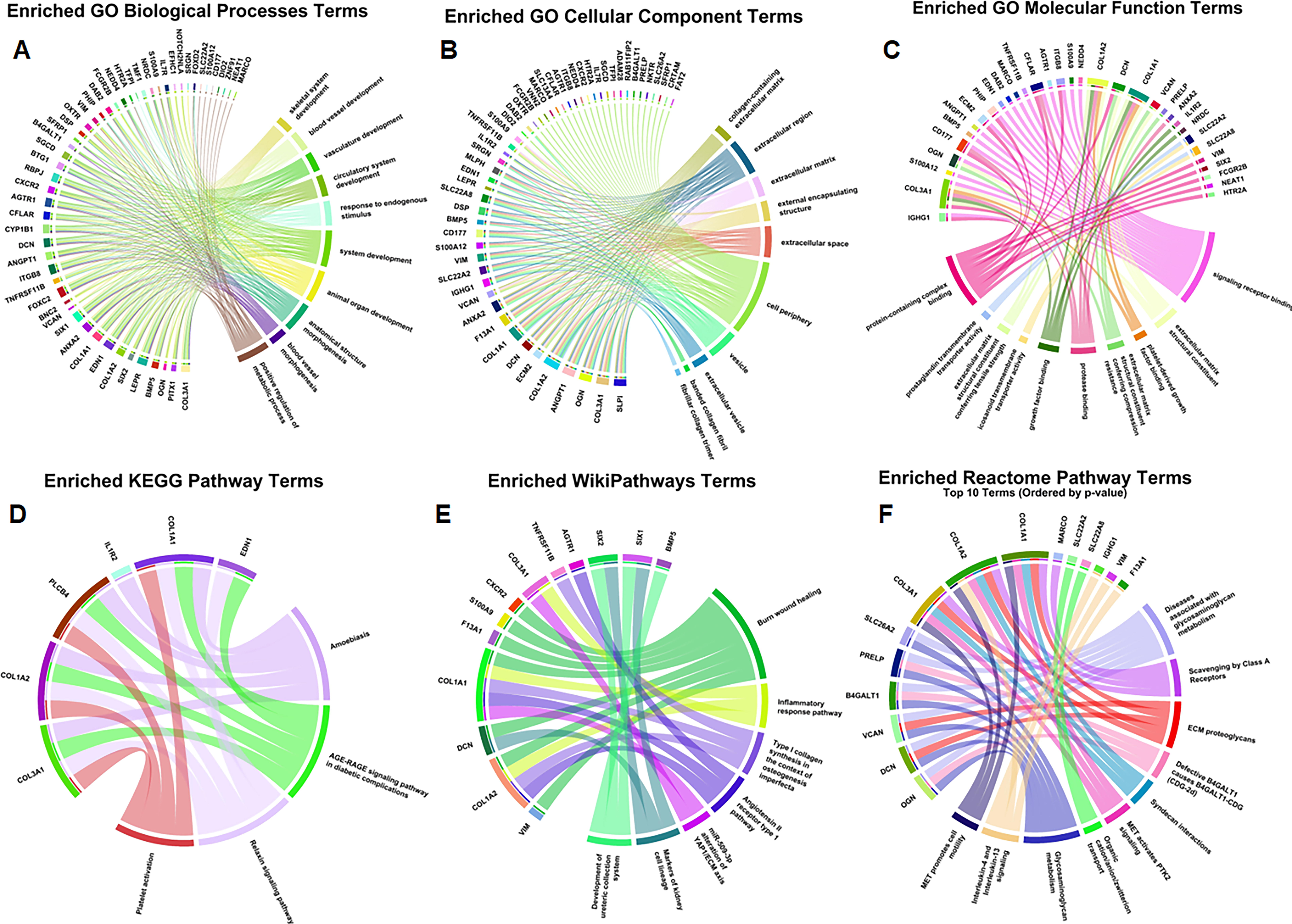
Circos plot for top 10 enriched GO terms
PPI network analysis and hub gene identification
Protein interactions in a network are useful in identifying core modulatory genes (Sidhu et al., 2003). To this end, we used the STRING database (https://stringdb.org/), which is a useful source for known and predicted protein-protein interactions (PPIs) (Mering et al., 2003). For the construction of the PPI network of the 80 identified DEGs, we used the StringApp tool in Cytoscape with a confidence score of ≥0.4 (Fig. 4). From the network, singleton nodes were removed to increase clarity and the network’s area of focus, as shown in Figure 4A. To find out and analyze the major hub genes of the PPI network, we used the cytoHubba plugin in Cytoscape with basic settings. Our strategy was to use several topological measures for the same reason, as different algorithms may reveal the node’s centrality in the network. We ranked genes based on seven different algorithms from cytoHubba, including Degree, MCC, MNC, and centrality measures based on shortest paths, such as BottleNeck, Closeness, Stress, and Betweenness. The outcomes of these algorithms applied for hub gene identification are visually represented in Figure 4B–H. Notably, we observed overlapping results across the seven topological algorithms.
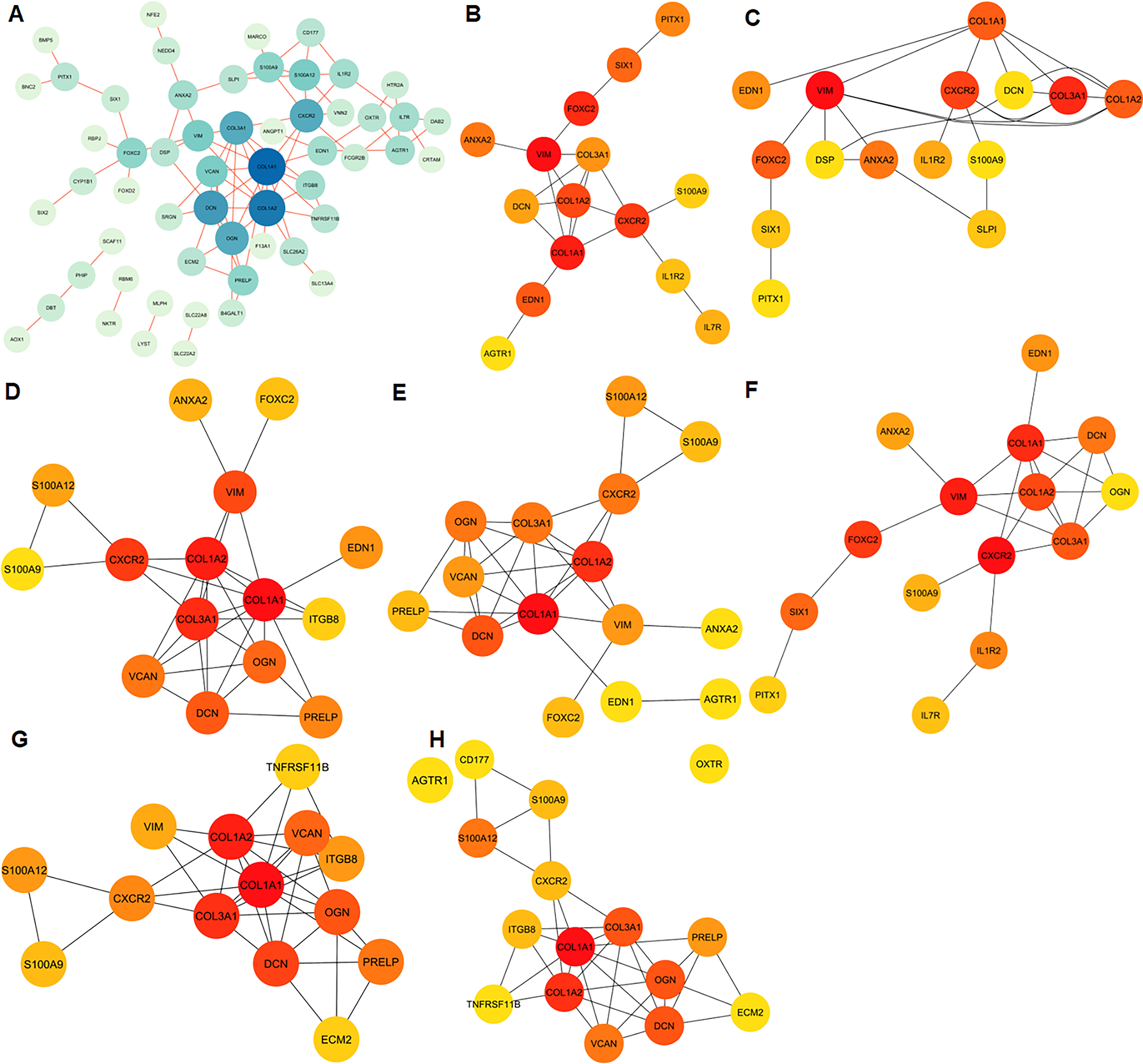
Protein-protein interaction (PPI) network analysis and hub gene identification
Further, to identify the most influential hub genes, we employed the UpSetR package (Conway et al., 2017) to visualize the intersections of these algorithmically ranked genes, as illustrated in Supplementary Figure S3. Ultimately, this rigorous analysis identified seven hub genes: COL1A1, COL1A2, COL3A1, DCN, CXCR2, S100A9, and VIM. These genes emerged as central players within the PPI network, signifying their potential significance in the context of the studied biological system, i.e., HD.
Discussion
HD is a complex and progressive neurodegenerative disorder that affects the individual’s motor control and brings about severe complications in terms of diagnosis and therapies (Sampaio, 2024). We analyzed several datasets to investigate the mRNA aspect of the HD molecular process. We aimed to advance the process of identifying diagnostic biomarkers and therapeutic targets for this disabling disease. First, we identified 80 DEGs associated with HD. Out of these genes, 20 were upregulated and 3 downregulated in both studies and passed through stringent statistical criteria. This observation has further emphasized the fact that there is a change in transcription that occurs in HD and which genes are affected. Among the identified genes, the most notable one was PITX1, which was found to have a high FC in its expression. PITX1 is a homeobox transcription factor that has significant functions in forming multiple tissues, particularly the nervous system (Poulin et al., 2000; Prince et al., 2011). These findings indicate that its upregulation in HD may be involved in disease pathogenesis and may affect neuronal development or function. Another candidate gene that was found to be upregulated was IGHG1, which is an immunoglobulin that is involved in immune response (Larsson et al., 2020). Its upregulation might be associated with the immune process that takes place in the brain, and it is known to be involved in neurodegenerative diseases (Chowdhury et al., 2020). Thus, it is possible that understanding the involvement of immune processes in HD will open new possibilities for its treatment.
We performed GO enrichment analysis to gain insights into the biological functions of the DEGs. Several enriched GO terms in the category of biological processes stood out. “Skeletal system development,” “blood vessel development,” and “vasculature development” were among the top enriched terms. These processes are important in HD pathogenesis as they point to some of the possible functions of DEGs in tissue formation, regeneration, and remodeling. These findings are of particular interest because HD is linked to neuronal degeneration and changes in the tissue structure. The use of the enriched molecular function terms helps in understanding the molecular processes associated with the DEGs. “Signaling receptor binding,” “extracellular matrix structural component,” and “platelet-derived growth factor binding” suggest that DEGs are involved in signaling and in structural and growth factor roles within the extracellular matrix. These functions may be associated with cellular signaling, the process of tissue reorganization, and neuroinflammation, which are all involved in the development of neurodegenerative diseases.
Certain pathways that are affected by the DEGs include amoebiasis, the AGE-RAGE signaling pathway in diabetic complications, and the Relaxin signaling pathway. These pathways could be useful in explaining the cause of the disease. This is why the “Amoebiasis” pathway is quite interesting, since it deals with host reactions to parasitic diseases. The identified pathways, like the AGE-RAGE signaling pathway and extracellular matrix organization, indicate that metabolism dysfunction may be associated with neuroinflammation in HD. These results corroborate the prior studies which suggest that altered signaling pathways are involved in the neuronal death and dysfunction in HD and disrupt cellular stress response pathways.
While HD is not a parasitic disease, this pathway’s activation could indicate an aberrant immune response in HD or alterations in the gut-brain axis, which has been suggested to play a role in neurodegeneration (Westfall et al., 2017). The “AGE-RAGE signaling pathway in diabetic complications” pathway connects HD to metabolic and vascular processes. HD affects various metabolic pathways, and vascular changes have been observed in HD patients. This pathway’s enrichment may explain the interplay between metabolic dysfunction and neurodegeneration in HD. The “Relaxin signaling pathway” involves various physiological processes, including tissue remodeling and inflammation. Its implication in HD suggests that tissue integrity and inflammatory responses may be significant factors in disease progression.
Constructing the PPI network and the identification of hub genes help in understanding the organizational structure of DEGs. The following seven hub genes were obtained: COL1A1, COL1A2, COL3A1, DCN, CXCR2, S100A9, and VIM. These genes are in the middle of the network and might be involved in the development of HD. Among the hub genes, collagen genes (COL1A1, COL1A2, and COL3A1) are of interest since they are the proteins of the extracellular matrix (ECM) (Christner and Ayitey, 2006). Changes in the integrity of the ECM have also been found to be linked with neurodegenerative diseases such as HD (Hernandez et al., 2023). These genes may be related to ECM and may play a role in the health and survival of neurons. DCN (Decorin) is another hub gene that has a possible relation to HD. Decorin is an extracellular matrix protein, and it has functions in tissue remodeling and inflammation (Stamenkovic, 2003). Its presence among hub genes signifies that these processes are crucial in HD.
CXCR2 is a chemokine receptor that plays a role in immune responses and inflammation (Liu et al., 2014). Its involvement suggests that neuroinflammation may be a significant aspect of HD pathogenesis (Veenstra and Ransohoff, 2012). S100A9, a member of the S100 protein family, is associated with inflammation and immune responses (Zhao et al., 2012). Its presence among hub genes underscores the potential involvement of immune processes in HD. VIM (Vimentin) is an intermediate filament protein that maintains cell structure (Wunderlich et al., 2015). Its upregulation may reflect cytoskeletal changes in neurons affected by HD. Our findings align with previous transcriptomic studies in HD, where extracellular matrix-related genes (COL1A1, COL3A1) and immune response genes (S100A9, CXCR2) were implicated in disease progression (Meem et al., 2023; Pampuscenko et al., 2025). Similar hub genes have been identified in different large-scale omics analyses (Mastrokolias et al., 2015), reinforcing their potential as biomarkers and therapeutic targets.
Conclusions and Outlook
The present study has discovered seven hub genes and two top DEGs, IGHG1, and PITX1, as potential molecular targets of relevance to HD pathogenesis, and thus contributes to future efforts for translational omics research and precision diagnosis and treatment of HD. Understanding the molecular insights of HD is crucial for developing targeted therapeutics and diagnostic biomarkers. Experimental validation of the DEGs and hub genes in cellular and animal models of HD is essential to confirm their roles in disease progression. Additionally, investigating the potential crosstalk between the identified pathways and the contributions of immune responses, metabolic processes, and tissue remodeling to HD pathogenesis should be a priority. Our integrative analyses of mRNA profiles in HD have unveiled critical molecular insights into the disease in this regard. This study adds significant information on molecular insights on HD which may ultimately lead to improved outcomes for individuals affected. A better understanding of genomics, environmental and epigenetic factors (Brulé et al., 2025) will facilitate HD research in the future.
Footnotes
Acknowledgments
M.I.H. acknowledges the Council of Scientific and Industrial Research for financial support [Project No. 27(0368)/20/EMR-II]. AH acknowledges the Researchers Supporting Project Number (
Authors’ Contributions
A.H.: Data curation, validation, formal analysis, funding acquisition, writing—review and editing; T.M.: Conceptualization, investigation, data curation, validation, writing—original draft; S.K.: Data curation, writing—review and editing, funding acquisition, investigation, validation, M.F.A.: Data curation, validation, funding acquisition, conceptualization, supervision, D.K.Y.: Writing—review and editing, supervision, Md. I.H.: Conceptualization, supervision, funding acquisition, project administration, writing—review and editing.
Data Availability Statement
The article contains all of the data created or used during the present research.
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
This work is supported by the Central Council for Research in Unani Medicine (CCRUM), Ministry of AYUSH, Government of India (Grant No. 3-69/2020-CCRUM/Tech) and the Researchers Supporting Project Number (RSPD2025R980) at King Saud University, Riyadh, Saudi Arabia.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.