
Abstract
With the renewed interest in phage research, coupled with the rising accessibility to affordable sequencing, ever increasing numbers of phage genomes are being sequenced. Therefore, there is an increased need to assemble and annotate phage genomes. There is a plethora of tools and platforms that allow phage genomes to be assembled and annotated. The choice of tools can often be bewildering for those new to phage genome assembly. Here we provide an overview of the assembly and annotation process from obtaining raw reads to genome submission, with worked examples, providing those new to genome assembly and annotation with a guided pathway to genome submission. We focus on the use of open access tools that can be incorporated into workflows to allow easy repetition of steps, highlighting multiple tools that can be used and common pitfalls that may occur.
Introduction
The recent renaissance in phage research and developments in sequencing technologies has resulted in increasing numbers of phage genomes being sequenced. 1 While phages are similar to bacteria in how they can be assembled and annotated, there are significant differences. The most important difference being the expectation for phages that their genomes can be completely assembled with short reads. Many tools developed for bacterial genome assembly and annotation work well with phage genomes, with some modifications.
Here we give an overview of phage genome assembly, structural and functional annotation, and submission to a member of The International Nucleotide Sequence Database Collaboration. We provide examples, highlighting the choices that can be made with a focus on open-source software via a Linux interface (Supplementary Data S1), which allows the process to be streamlined by the production of a workflow manager or simple bash script. Details of scripting and workflow managers are not discussed in detail other than to say tools such as NextFlow 2 and SnakeMake 3 allow the adaptation of pipelines written in bash and other scripting languages. There are also workflows available via web browsers that offer all in one solution such as PATRIC and the Galaxy interface 4 for phage genome assembly.
The stages of bacteriophage assembly and annotation are split into three areas: genome assembly, genome annotation, and genome submission. Each of these sections is broken down into subsections with suggestions on tools and pitfalls that might occur at each step. The process covers the questions raised by Turner et al.
While it might appear counterintuitive, we recommend those new to sequencing phage genomes to look at the requirements for genome submission before any genome sequencing. Understanding the data required for genome submission before starting the process can save considerable time when the time comes for final submission. For sequence submission to the European Nucleotide Archive (ENA), the current information that is required to gain an accession number is the following: a taxonomy ID, a unique locus tag, phage name, project number, depth of coverage, library insert size, sequencing platform, and assembly software. Collating this information as the assembly and annotation process is completed can save considerable time when genomes are to be submitted.
Bacteriophage Genome Assembly
Presequencing
Before any genome assembly, it is important to determine in advance what is required from the final genome assembly. Bacteriophages can have different packaging strategies that will result in different genomic termini or circularly permuted genomes. 5 If it is important to know the exact termini of the genome, then the choice of sequencing is a critical factor and deciding this beforehand can prevent further experimental work later. There are many different library preparation methods for both short- and long-read sequencing.
The choice of library preparation method will be based on several factors, including the amount of input genomic DNA (particularly high for long-read sequencing), provision by service providers, the number of genomes to be sequenced, and cost. The use of transposon-based library preparation methods such as NexteraXT that uses tagmentation has the benefit of requiring minimal amounts of input DNA and providing easy automation for multiple library preparations simultaneously. 6 However, the insertion of a transposon requires some bases upstream of the insertion site, and thus, the terminal bases will never be sequenced. 6 This prevents easy identification of the termini based on sequencing data alone if transposon-based approaches are used as tools, such as PhageTerm, not compatible with such library preparations. 7
The vast majority of bacteriophage genomes can be sequenced via the use of short-read sequencing if they are sequenced as individual isolates. 8 With limited advantages of assembly, it is possible to use longer reads as most phages isolated to date do not have repeats that prevent assembly, as can happen with large bacterial genomes. 8
However, the use of long-read technologies such as the Oxford nanopore technology (ONT) and PacBio offers other advantages including identification of DNA modifications, 9 although these techniques are far more specialized and identification of base modification is covered here. ONT sequencing via the use of a minION does offer other advantages compared to Illumina sequencing, including rapid turnaround time from DNA to genome sequence, sequencing a single genome at a time and allowing users to sequencing within their own laboratory with minimal investment in infrastructure. 10 However, the initial genome assembly of ONT reads can be more challenging than that of Illumina sequencing. For the focus of this overview, we focus on the use of short-read Illumina sequencing and long-read nanopore sequencing.
Genome sequencing
To obtain an assembly, it is necessary to obtain reads from a phage genome. For dsDNA phages, it is now apparent this is not always a trivial process. It has been known for decades that phages can modify the nucleotides in their genomes. 11 However, there are now increasing reports of phages with hypermodified DNA that is recalcitrant to standard sequencing methods.12–14
To overcome this, innovative approaches of using RNA-Seq to reconstitute the genome from phage transcripts 14 or rolling circle amplifications to remove DNA modifications have been applied.12,13 As the number of phages isolated increases and therefore sequenced, it is likely these approaches will have to be applied more often. For the purpose of this work, it is assumed that the genomic material is dsDNA from phages capable of producing virions and not recalcitrant to standard sequencing approaches.
Genome Assembly
Quality control of reads
The first step in the assembly of phage genomes is the quality control of read data and understanding how data have been produced and/or returned by a sequencing provider (Steps 1–3 in Supplementary protocol). If the sequence provider also provides an assembly of the data, it is desirable to understand how much data were used for the assembly. It is entirely possible to sequence a phage genome at >2,500 × coverage for ∼£100. Having an indication of the depth of sequencing is essential for optimal assembly. Assuming raw reads are provided, an initial assessment of read quality is an essential first step to produce optimal assemblies.
The use of FASTQC allows an overview of quality statistics of all the reads that have been returned and the number of reads per sample. 15 Following examination of the quality statistics, sequencing adapters and low-quality bases can be trimmed off. Reads can be trimmed with various tools, including but not limited to trim_galore, 16 sickle, 17 and BBDuk. Within this example, we utilize the Seqtk package (Step 4 in Supplementary protocol). Given that phage genomes are generally sequenced to a very high coverage, it is recommended to be strict in the parameters that are used for quality control of the data.
Having removed adapters and trimmed off poor-quality bases and removed any control DNA spike-ins that are used with Illumina sequencing (e.g., PhiX control), the next step is the assessment of how much data are present (Step 3 in Supplementary protocol). While more data might intuitively be thought to produce better genome assemblies, this is often not the case with short-read data. Using 30 × coverage allows the assembly of most phage genomes and using very high coverage of data can result in assembly problems 8 Therefore, it is suggested that reads are randomly subsampled to give ∼50–100 × coverage of the genome. Given that genome sizes are rarely known before assembly, this can be roughly calculated using the number of reads, read length, and an “average genome” size.
(number reads × read length)/expected genome size
For example, MiGs (https://www.migscenter.com/) return on average 450 Mbp of data per genome, which is 15,000,000 paired-end reads of 150 bp in length, equating to 4,500 coverage of a 100 kb phage genome. Reads can be randomly subsampled with a variety of tools, including Seqtk toolkit, 18 BBnorm, or reformat.sh as part of the wider BBtools package. Within this example Seqtk was used (Step 4 in Supplementary protocol).
The process of quality control is different for ONT data, with raw output files being FAST5 rather than FASTQ sequences. Initially base calling is required to convert FAST5 to FASTQ, followed by demultiplexing (if multiple samples were combined in a library), which can be performed with Guppy. Further demultiplexing can be done if necessary with a variety of tools including Porechop 19 and Qcat. 20 Overall read metrics can be gained using various tools including Poretools 21 and MINIOQC. 22 There are extensive tutorials available through the nanopore community portal, providing guidance on nanopore tools.
Utilizing nanopore sequencing will generally require increased read depth compared with Illumina data. With limited publications to date using minION data for sequencing phage isolates, the optimal read coverage is unknown. Based on data available for bacterial genomes, it is likely a sequencing depth of 100–200 × will be required to assemble high-quality genomes, 23 with consensus base calling accuracy of >Q50 (99.999% accuracy) (https://nanoporetech.com/accuracy). While it is possible to assemble phage genomes from far lower coverage, these will likely contain numerous base calling errors, which may lead to the presence of artificial frameshifts within coding sequences. Further details on different assembly options using nanopore data can be found in Wick et al. 24
It is also possible to assemble phage genomes using a hybrid approach, combining long and short reads. For the sequencing of complex phage communities (viromes), this approach is advantageous.25,26 However, for individual phage isolates, the approach has minimal benefits and will add considerable costs and thus not recommended for the majority of phage genome sequencing projects.
Assembly
There are numerous genome assemblers suitable for phage genomes, including but not limited to SPAdes, 27 Megahit, 28 Ray, 29 and MetaVelvet. 30 Previous research found that most assemblers produce consistent genome assemblies. 8 We recommend the use of SPAdes with the option “–only-assembler,” based on previous analyses. 8 If for some reason the input DNA was amplified before sequencing (e.g., to remove DNA modifications or meet input requirements), then the “–sc” flag for multiple displacement amplifications is advantageous. With SPAdes, the output file “contigs.fasta” will contain the resultant assembly that may contain a single contig or multiple contigs (Step 5 in Supplementary protocol). Other assembly programs will produce very similar outputs but likely have a different output name.
There are numerous assemblers for ONT reads, including SPAdes, 27 FLYE, 31 wtdgb2, 32 Unicycler, 33 and Tricycler. 24 Given the relatively small number of phage genomes sequenced to date with this technology and rapidly evolving software, a consensus on the most appropriate assembler has not been reached for phages. The assembly with any of these tools may require some optimization of parameters as detailed for each tool. The following assembly may also require rounds of polishing to correct errors, which can be achieved using a variety of tools including Medaka 34 and Pilon. 35 The tool FLYE was used as an example in this work (Step 17 in Supplementary protocol). Once a genome has been assembled, the annotation steps are the same regardless if the genome was assembled from Illumina or ONT reads.
Assembly validation
At this stage, it is important to check the assembled contigs to help determine how well the assembly has worked and identify any errors. If SPAdes was used, the contigs.fa output file will contain the contigs of interest (Step 5 in Supplementary protocol).
Preliminary checking of contigs
It is most likely that the resulting contigs from the assembly will contain a single contig with high coverage, while the remaining contigs are very short with low coverage, this is indicative of assembly of a complete phage genome. Variations from this will require some further work to identify what they are. Contigs from contaminating host DNA need to be removed before phage genome annotation. Assemblies can be assessed by the use of Bandage 36 that allows the assembly-graphs to be visualized. The input file for Bandage is an assembly graph file rather than a FASTA file; if SPAdes is used for assembly this will be a *.fastg file.
In conjunction with genome coverage, the assembly graph can be used to view the assembly and to identify common assembly-associated issues (Steps 6–8 in Supplementary protocol). When viewing the assembly graphs, it is important to note that while phage genomes may appear circular, this does not mean that the genome is circular. The circularity arises as an artifact either from a circularly permuted genome or terminal repeats making it appear circular (Fig. 1, see Q4 in Turner et al.).
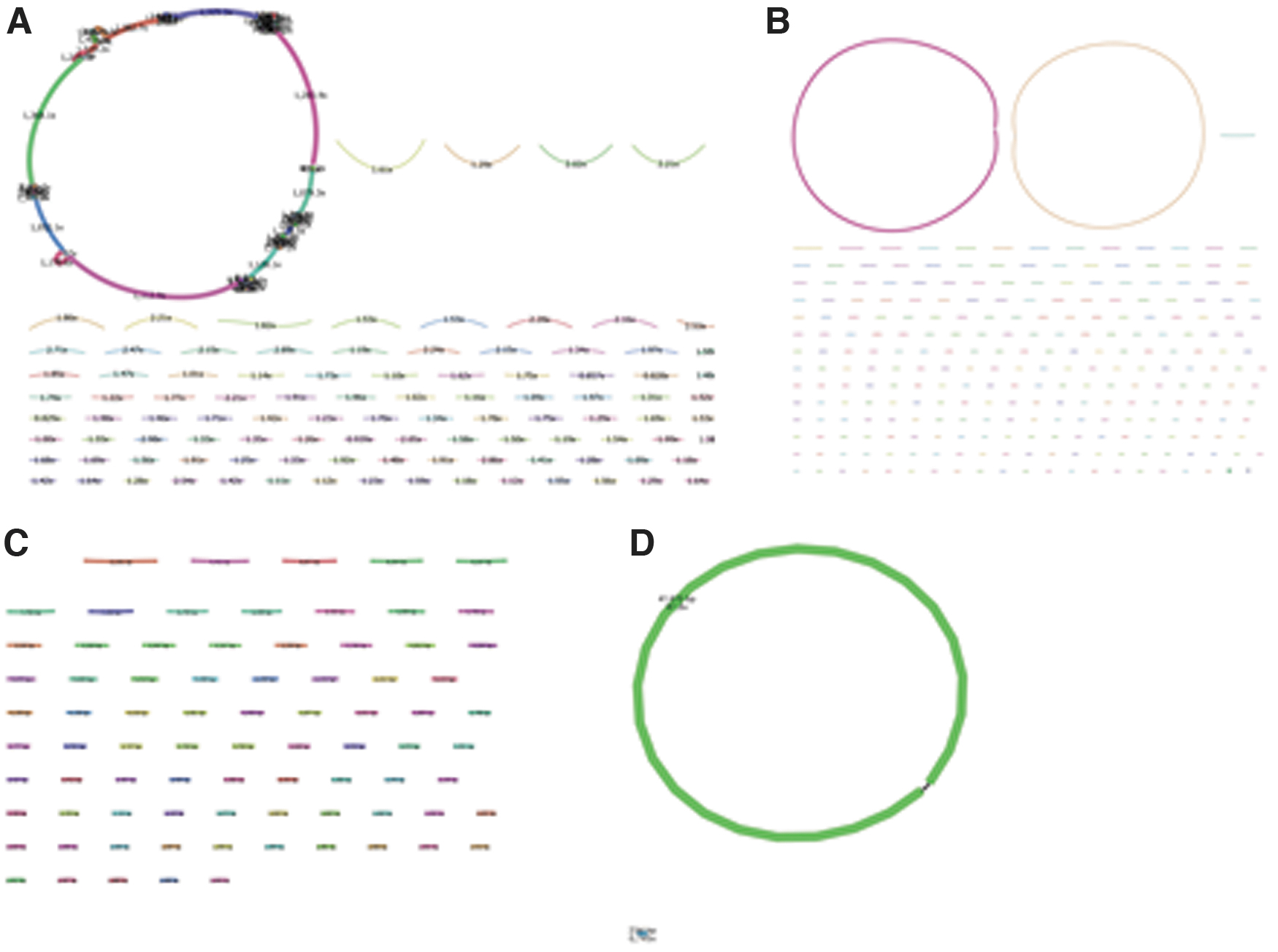
Depth of sequencing coverage per contig
The absolute read coverage per contig can be obtained by mapping reads to a set of contigs. There are multiple tools to map reads, including BWA-MEM, 37 Bowtie2, 38 bbmap.sh, 39 and minimap2, 40 all of which are suitable for mapping, and coverage can be calculated using other tools. Using bbmap.sh allows read coverage of each contig to be directly calculated as an output file with the “–covstats” flag, without the need for another tool (Step 7 in Supplementary protocol).
The resultant output file allows coverage for each contig to be assessed, which is a useful parameter to assess how well the assembly has worked. In most cases, this consists of a single large contig with high coverage and smaller contigs with far lower coverage. This is indicative of a complete phage genome with some background host DNA. If the coverage is very high >200 × , further subsampling of reads is recommended. Alternatively, if a low coverage (<20 × ) of the largest contig is obtained, subsampling and keeping a higher proportion of reads are recommended.
Based on the results obtained, it might be necessary to resample the reads and repeat the assembly process. This may well be an iterative process requiring multiple subsamplings and assembly runs (see Q1 in Turner et al.).
If reads were first subsampled, it is also good practice to map all the short reads that passed quality control, providing statistics of depth of sequencing coverage for the sample, which is required later for genome submission and can be used for error correction with pilon. 35 Reads that do not map to the putative phage contig can also be collected at this stage. The rationale for doing this is to check if other phages are present. For some bacterial hosts, low-level induction of prophages can occur, and this will not have been detected in the reads that have been subsampled and assembled.
The removal of reads associated with the most abundant phage contig increases the ability to identify any prophages that have been induced at a low level in a subsequent assembly. To do this, all reads that were not mapped to the putative phage should be extracted and reassembled with SPAdes (or any other assembler).
Preliminary identification of closest relatives
To aid in genome annotation, it is useful to identify if the putative phage genome is similar to previously sequenced phages (Step 9 in Supplementary protocol, see Q3 in Turner et al.). There are multiple ways to identify close relatives, including the INPHARED database and associated scripts, which provide a command line interface to rapidly compare against complete phage genomes in GenBank. 1 Alternatively, BLASTn against the Virus database can be utilized to search via a web interface using the taxaID Viruses: 10239 to search only against viruses.
The similarity to other phages and the sequencing library method used determines the next steps in annotation. This also helps early on to determine if a complete phage genome has been assembled, as any closely related phage genome would be expected to have a similar genome length. The recent development of PhageClouds offers a web interface to rapidly compare against a database of >600,000 viruses with a visual representation of genome size that helps judge completeness based on similarity and size of other phages. Alternatively, a prediction of genome completeness can be assessed using the command line tool checkV, which also indicates termini type 41 (see Q2 in Turner et al.).
Genome reordering
It is important to note that assemblers do not output genomes that are ordered correctly; this does not mean the assembly is wrong, rather it may require reorientation as it is not designed to identify terminal repeats or identify circularly permuted genomes (see Q4 in Turner et al.). How reordering is approached will depend on the library preparation method and how related to other phages the new phage genome is (Fig. 2).
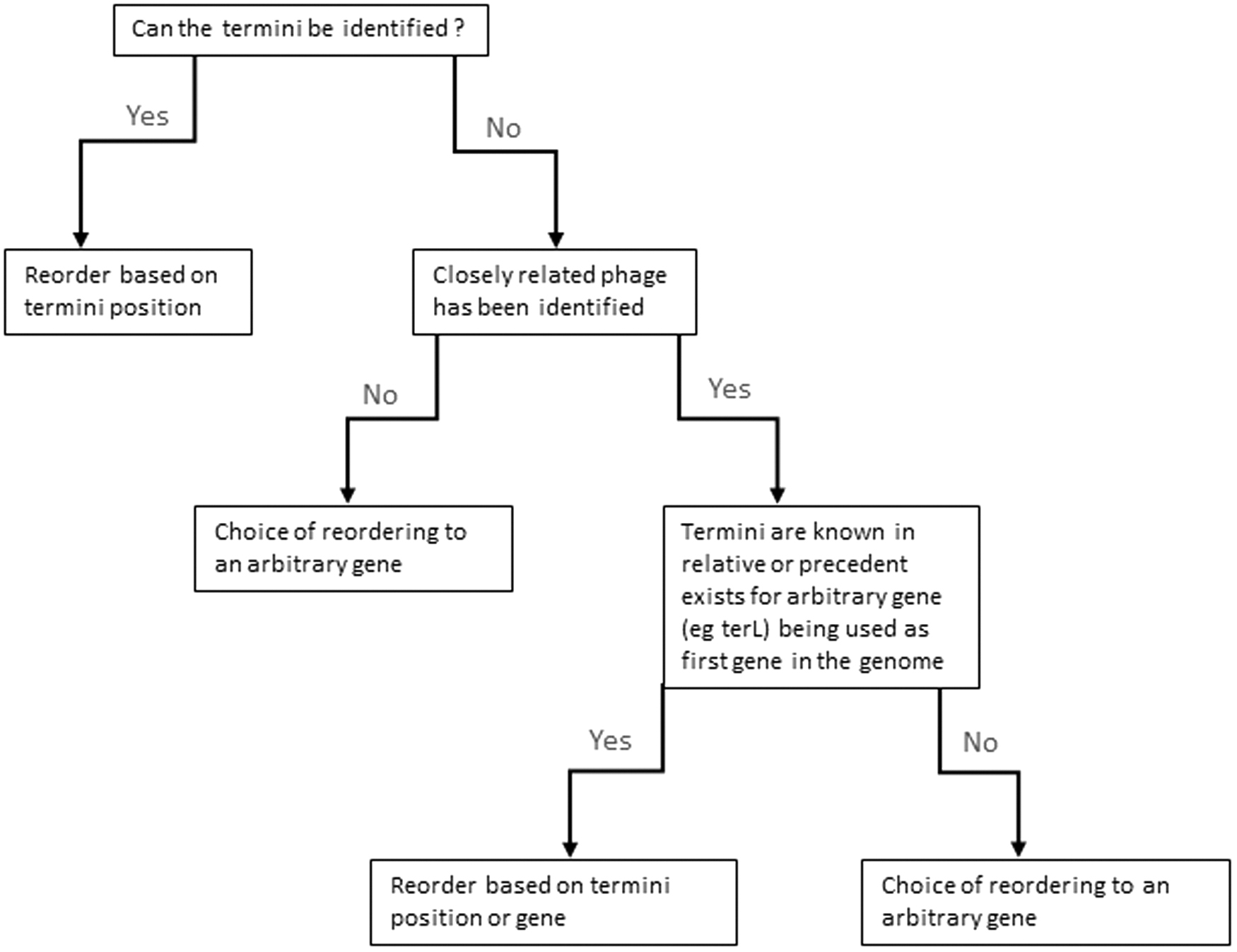
Decision process for reordering of phage genomes.
If a nontransposon-based library preparation was used, then PhageTerm can be used to predict the packaging strategy of the phage and automatically reorder the genome if it has defined termini. 7 Alternatively, if a transposon-based approach was used, other approaches will be required. If the preliminary phage identification resulted in it being similar to other phages (>95% ANI), it is possible to use the information from a close relative for genome reordering, if the termini of a relative have been identified previously. The outputs of CheckV also identify terminal repeats within sequences (see Q2 in Turner et al.). Otherwise, a specific gene may be arbitrarily selected as the start point of close relatives (e.g., terL, split between terS and terL [see Q2 in Turner et al.]). Utilizing this gene and maintaining consistency across similar genomes have some benefits of visualizing genomic comparisons.
Previously, it has been suggested that the large terminase subunit should be set as the first gene, if termini have not been identified. 42 However, the benefits of this are limited if the phage is not closely related to any other phages. It is important to state if the termini have been identified and, if not, to ensure that the arbitrary start point is not intragenic.
Genome reordering can be achieved manually by first identifying the new first base in the genome and manually editing the sequence, so any bases upstream of the new first base are moved to the 3′ of the genome (Fig. 3). If the genomes are closely related, Mummer4 allows rapid whole-genome alignments. 43 Whole-genome alignments of very closely related phages can be achieved with other tools such as MAFFT 44 or MAUVE. 45
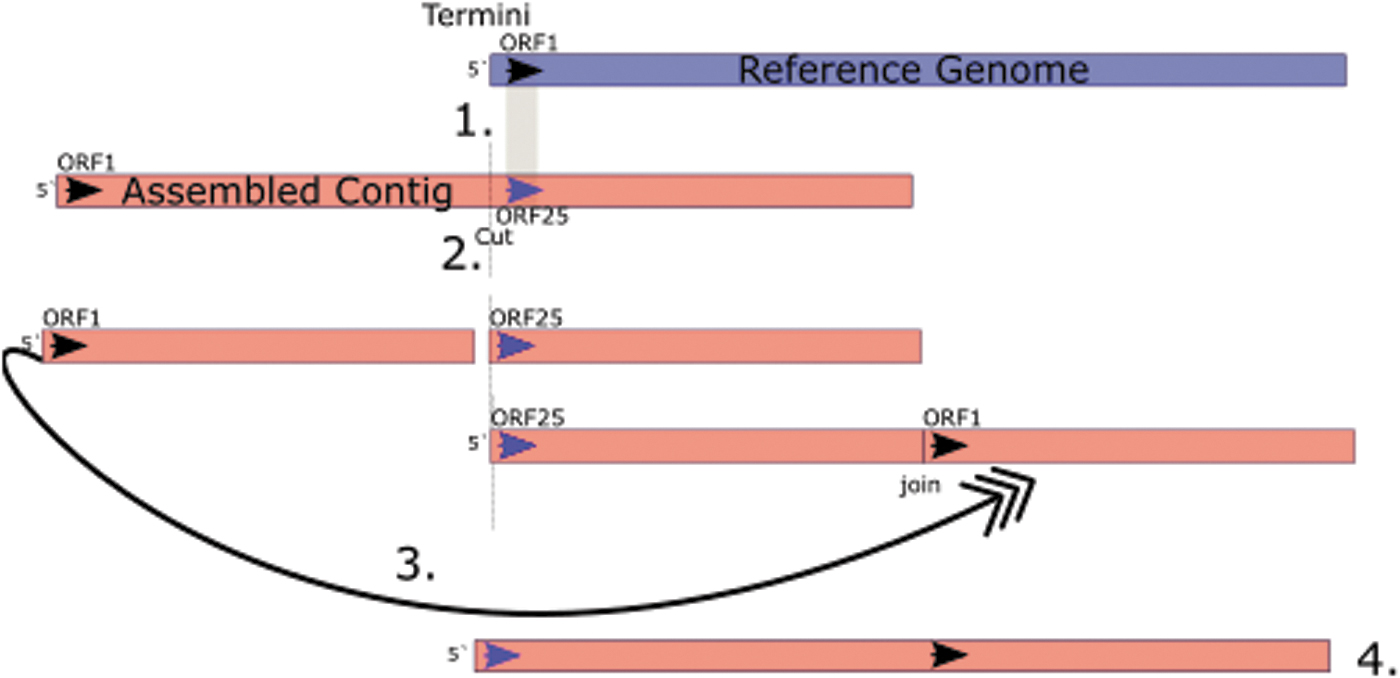
Reordering phage genomes. (1) Align the genome against a reference genome and identify the gene/start point that will become the new start position. (2) Cut the genome upstream of the position that will become the new 5′ start. (3) Move the cut fragment to the left of the new start position to the 3′ end and join. (4) Check for assembly errors with Pilon and recall all genes.
Alternatively, an initial annotation can be performed to identify the gene that will be used as the arbitrary start of the genome, for example, terL. Ordering the genome at this point can save considerable time later. Whenever a genome is reordered and before final gene calling, the use of plion 35 or REAPR 46 is advised to ensure no errors have been introduced and correct any local mis-assemblies along with erroneous base calls in the original assembly. 35
Troubleshooting genome assembly
While most phage genomes will assemble, there are always those that do not. There are several reasons for incomplete assembly, a common reason being very high or low read coverage (Table 1). Thus, it is recommended to subsample reads before assembly. In addition, there are other reasons that a single-phage contig is not assembled. Including bias in mol GC% content, with transposon-based library preparations well known to target AT-rich regions or undercut high GC regions that can lead to broken assemblies across these regions. 47 Alternatively, the presence of multiple very similar phage genomes with high microdiversity can cause assemblies to break. 48 Some of these issues can be resolved bioinformatically by viewing assembly graphs (*.fastg from SPAdes output) in Bandage and the use of polymerase chain reaction to sequence any gaps in the genome.
Common Results Obtained from Sequencing Data
Genome Annotation
Gene calling (structural annotation) (see Q5 in Turner et al.)
There are multiple stand-alone tools for gene calling and the prediction of tRNAs. For gene calling, Prodigal, 49 GeneMarkS, 50 Glimmer, 51 and MetaGeneAnnotator 52 are commonly used and are available through a command line interface or web interface. Recently, a phage-specific gene calling algorithm has been developed (Phanotate) that incorporates several of the above tools to identify a consensus from multiple tools. 53 For the identification of tRNAs, Aragorn, 54 tRNAFinder, 55 or tRNAScan-SE 56 can be utilized. If these tools are run alone, the results must be collated into a single coordinate file to overlay the information onto a genome using tool such as Artemis 57 or Ugene. 58
Artemis and Ugene also allow users to manually BLAST each sequence to provide a functional annotation or export all predicted gene sequences to allow a batch BLAST analysis (see Q6 in Turner et al.).
Alternatively, Prokka can be used that combines the process of coding sequences, annotation of genes, and formatting of files for submission to an International Nucleotide Sequence Database. Prokka has considerable advantages as it allows rapid and consistent annotation of a genome with formatting of files ready for submission with minimal reformatting. Prokka also provides files that can be directly used as input for downstream genome analysis. Gene calling in Prokka is provided by Prodigal, with tRNA identification through ARAGORN. 54
In addition, CRISPR arrays and tRNAs/tmRNAs are searched for by default. The inbuilt databases are primarily designed for bacteria, but are easily adapted for bacteriophages. Prokka prescribes a function through a hierarchical approach, using a BLAST database, followed by more sensitive Hidden Markov models (HMMs). For the more experienced user, a series of scripts can be utilized to create a specific database from a set of genomes, for instance, all current phage genomes. It also allows addition of HMM databases, allowing the use of prebuilt databases such as the pVOG database 59 or PHROGs. 60
The use of PHROGs in particular provides good initial functional annotation of phage proteins. Alternatively, using the “–proteins” option allows the user to provide a GenBank file used for annotation, which is particularly useful if a closely related phage is already well annotated (Steps 13–15 in Supplementary protocol).
The output of Prokka provides a good starting point for further annotation and eventual submission to a database. The GenBank file can be read directly into graphical interfaces such as Artemis or Ugene for further annotation and manipulation or can be manipulated directly with a text editor. Often the automatic annotation will result in uninformative product descriptions, for example, “gp23,” which is ambiguous and needs correction to describe a function such as “major capsid protein” (see Q7 in Turner et al.). The choice of database used for annotation can have significant effects on the number of proteins that are assigned a potential function.
Annotation of virion structural genes
With increasing amounts of phage genomic data, machine learning approaches have been utilized to make predictors for specific gene types. Machine learning was utilized to distinguish between structural virion and nonvirion proteins, although successful was not easily implemented into pipelines. 61 Recently, more user-friendly approaches have been developed to classify phage structural proteins that allow access via a web server (http://edwards.sdsu.edu/phanns) or integration into pipelines via the command line. 62 For phages that have limited similarity to well-characterized phages, this can help considerably in ascribing predicted gene function.
Template-based homology searching
Further predicted protein functions can be gained using comparison of phage proteins with known protein structures, through a process of template-based modeling. This approach is available through the Phyre2 website (www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) that allows users to submit individual sequences or batches of jobs. 63 Given the computational cost and time required, there is limited benefit of running Phyre2 analysis on well-annotated proteins such as DNA polymerase and there is more benefit for proteins that do not have any form of functional annotation initially.
Specific annotation for therapeutic phages
For phages considered for use in therapy, there is considerable interest for determining whether they are strictly lytic and if they carry antimicrobial resistance genes or genes that might alter their bacterial host virulence (see Q8 in Turner et al.). The presence of antibiotic resistance genes in lytic phages is a rare event,1,64 while carriage of antibiotic resistance genes and virulence genes in temperate phages is more common. 1
The software ABRicate allows users to rapidly search against a range of antibiotic resistance gene and virulence databases for such genes. 65 Alternatively AMRFinder (https://www.ncbi.nlm.nih.gov/pathogens/antimicrobial-resistance/AMRFinder/) or graphical interfaces to the CARD database (https://card.mcmaster.ca/) 66 and the Virulence factor database (www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi?func=VFanalyzer) are also available. 67 Careful use of thresholds should be used for identification of such genes1,64 to avoid over interpretation (see Q8 in Turner et al.).
Prediction of lytic and temperate lifestyles
There are many tools available for prediction of the lifestyle of phages, and in the case of phage therapy, this is particularly relevant where the avoidance of temperate phages is preferred (see Q8 in Turner et al.). These include BACPHLIP, 68 PhageAI, 69 and PHACTS, 70 all of which offer web interfaces for prediction with stand-alone code available for PHACTS and BACPHLIP to allow integration into pipelines with application programming interface access to PhageAI.
More recently, tools that simultaneously predict the lytic nature of a phage and the presence of antibiotic resistance genes (ARGs) or virulence factors have been developed for example, PhageLeads (http://130.226.24.116/phagecompass/index.html#/PhageLeads). As with all tools, these are only predictions of the life cycle that require experimental validation. The recent characterization of the cyanophage S-TIP37 highlights that the presence of an integrase gene does not mean a phage will enter a lysogenic life cycle. 71
Introns and Inteins
Since the discovery of an intron in the thymidylate synthase gene of phage T4, 72 there have been increasing numbers of reports of introns in a variety of genes in phages, including ribonucleotide reductase, 73 terminase subunits, 74 DNA polymerase, 75 and the core photosynthesis gene psbA. 76 Current tools for gene prediction in phages will not correctly predict the presence of introns and therefore will require manual inspection. A telltale sign of an intron is the annotation of two genes that are adjacent to each other with the same function, and this often results as the intron introduces a premature stop codon into the gene. By alignment of suspected intron containing genes with homologues lacking introns, it is often possible to identify the insertion site of introns through manual inspection of alignments (see Q5 in Turner et al.).
Inteins, which are autocatalytic internal proteins that are capable of excising themselves from the surrounding extein protein, are also found within phages. Similar to introns, they are often found in conserved phage genes such as those encoding ribonucleotide reductase, 77 DNA polymerase,78,79 and terminase subunits. 12 In common with introns, these will not be identified by gene calling software and will require manual identification and curation.
Genome submission
In the interests of open data sharing, it is good practice to submit genome sequences before peer review of any publication of the work. Through this work, we have tried to identify the information that is required for submission of phage genomes through the ENA, which has a submission guide (https://ena-docs.readthedocs.io/en/latest/submit/general-guide/webin-cli.html). For submission to GenBank or DDBJ, similar information is required, although the process will differ. We have included a checklist of requirements for ENA (Table 2). For submission to the ENA, the registration of a study is required to gain a project number (https://ena-docs.readthedocs.io/en/latest/submit/study/interactive.html). At this point, unique locus_tags can also be registered. Both raw reads and an assembled genome can be submitted. If the phage is new, it may require the registration of a taxaID.
Check List of Information Required That Should Be Obtained Before Genome Submission
To complete the submission of raw reads and an assembled genome, a manifest file will be required, which also requires a sample accession (requires registration), depth of sequencing coverage (calculated previously), assembly program, and sequencing platform (https://ena-docs.readthedocs.io/en/latest/submit/assembly/genome.html?highlight=manifest#chromosome-assembly). For submitting the annotated genome to the ENA, the General Feature Format (GFF) file from Prokka needs to be converted to EMBL format and a manifest file produced. A GFF file can be converted to EMBL with the GFF3toEMBL script 80 with a manifest file created in a text editor (see Q10 and Q11 in Turner et al.).
Comparative Genomic Analysis and Beyond
Taxonomic classification
Complete comparative genomic analysis encompasses a huge area of research that goes beyond the scope of this work. Here we provide a brief overview of software that is available for comparative genomic analysis. For taxonomic classification of phages at the level of species and genus, there are now clear guidelines from the International Committee on Taxonomy of Viruses (ICTV) based on nucleotide identity. 81 As stated previously, VIRIDIC 82 offers an automated solution for comparison of phage genomes and identification of genera and species based on the nucleotide sequence. Other tools can be used and have been detailed elsewhere. 81
Identification of relationships beyond the genus level requires a more in-depth analysis. The development of all versus all protein analyses offers identification of subfamily- and family-level classification. The development of vContact2 places new phage genomes into viral clusters based on a network graph. 83 vContact2 is available as a stand-alone tool or via web interface through iVirus, with preformatted databases also available as input. 1
Alternative methods are also available via online tools including VipTree, which builds on the original phage proteomic tree, 84 with a simple interface that allows uploading of phage genomes. 85 One disadvantage of this online version is that it only utilizes the RefSeq database for phages, which is not representative of total phage genomes. 1 The stand-alone version does allow custom databases, but is slightly more involved to run. Classification can also be achieved using VICTOR, which allows users to upload genomes. 86 While the underlying algorithm can be scaled for comparisons and placement of thousands of genomes, the current web implementation is severely limited by the number of genomes that can be uploaded.
Further classification can be achieved by phylogenetic analysis based on single genes or core genes. The output files from Prokka (*gbk files) have the advantage that they can be directly input into comparative genomic software. For core-gene analysis Roary 87 and/or GET_HOMOLOGUES 88 offer a relatively easy pathway into core-gene identification. In the case of Roary, the resulting core-gene alignment file can be directly used as input for IQ-Tree for further phylogenetic analysis.
The use of GET_HOMOLOGUES in combination with GET_PHYLO_MARKERS 89 offers a complete workflow for phylogenetic analysis based on core genes. The approach of aligning core genes is preferable to attempting to align the complete genomes, which is not a suitable method due to poor alignment and incorporation of intergenic regions into the underlying alignments. For the classification of phage above the genus level in particular, contacting the ICTV for guidance is recommended (see Q13 in Turner et al.).
Microdiversity
When sequencing a phage genome, although the phage will likely have been purified through several rounds of purification, the extracted DNA is from a population of phages. Obtaining very high-coverage genome sequencing provides the opportunity to identify variations (single nucleotide polymorphisms [SNPs] and indels) within the population. There are multiple tools for SNP and indel detection, including FreeBayes, 90 SNIPPY, and VarScan2. 91
Conclusion
There are multiple methods for genome assembly, all of which have pros and cons. Here we have focused on open-source software that can be integrated into pipelines. The initial installation of software comes with a time cost. However, once installed, many of the steps explained here can then be easily repeated or automated with minimal effort. By providing a walk-through example, we hope to encourage others to utilize such tools and provide a reference point for others to begin their phage genome annotation journey.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
A.M. was funded through UK Medical Research Council grant CLIMB-BIGDATA (MR/L015080/1). N.S. was funded through the FHF—Norwegian Seafood Research Fund (“Genomic epidemiology and Phage-based Prevention of salmon associated Pasteurella,” grant No. 901707).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.