
Abstract
Background:
Lytic phages have been considered as a solution to mitigate the emergence of multidrug-resistant bacteria. Nevertheless, finding phages capable of targeting a broad host-range remains a significant challenge.
Materials and Methods:
Our study introduces two lytic phages isolated from hospital effluent, which are active against extended-spectrum cephalosporin-resistant Klebsiella pneumoniae.
Results:
Overnight coculture with host, two purified phage lysates yielded around 3.0 × 107 PFU/mL with an average 0.8 ± 0.2 mm diameter of clear, round, and non-halo plaques in both instances. The genomes of iPHaGe-KPN-11i (177,603 bp, 273 coding sequences [CDS]) and iPHaGe-KPN-12i (178,179 bp, 275 CDS) belong to the Pseudotevenvirus genus. Both phages have at least 120 genes with known functions, including 1 endolysin and 2 tRNAs, and are capable of lysing at least 12 distinct bacterial species in vitro.
Conclusions:
Most phages are host-specific, whereas our phages can kill multiple bacterial species, enabling their potential use for a broad range of hosts.
Introduction
Bacteriophages (phages) are naturally occurring viruses that specifically infect bacteria and were used to treat infectious diseases such as dysentery before the discovery of antibiotics. These viruses specifically target bacteria and typically do not directly interact with eukaryotic cells or archaea. Once infected, lytic phages harness the bacterial cell's resources to replicate and ultimately burst the bacterium open, releasing their progeny to pursue new bacterial prey. This mechanism of infection has made such phages a potential therapeutic tool.
However, with the advent of penicillin, the research spotlight shifted away from phages as antibacterial agents. In recent years, antimicrobial-resistance (AMR) has emerged and multidrug-resistant (MDR) bacteria have brought the spotlight once more on phages, prompting researchers to re-evaluate their therapeutic potential. 1
In Bangladesh, practices such as over-the-counter antibiotic sales, improper dosing, incomplete treatment courses, and the use of generic antibiotics have become commonplace. Unfortunately, these practices are fueling the development of resistant bacteria, which are increasingly detected.2–4 Just as with other low- and middle-income countries, Bangladesh is grappling with the emergence of MDR bacteria. Even the International Centre for Diarrheal Disease Research, Bangladesh (icddr,b), the world's largest cholera hospital, is not immune to this challenge.5,6
Ongoing AMR surveillance at icddr,b reveals that >90% of MDR pathogens in the community and hospitals are Escherichia coli and Klebsiella pneumoniae. Comprehensive characterization of a large collection of isolates (n = 3000) at the icddr,b Genome Centre (iGC) confirms the diversity of these pathogens, with 156 multilocus sequence types (MLST) of K. pneumoniae identified.
K. pneumoniae is one of the six difficult-to-treat pathogens known as ESKAPE—a group that also includes Enterococcus faecium, Staphylococcus aureus, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species. With the emergence of MDR K. pneumoniae strains, the World Health Organization has placed it on its critical priority list, highlighting the urgency to tackle infections caused by this pathogen given the limited antibiotic options available.
Beyond its resistance traits, the pathogen is notably virulent, adept at invading and enduring within hosts.7,8 The foremost virulence factor that ensures the survival of K. pneumoniae is the presence of capsular polysaccharides, which not only aid the bacteria in the evasion of the host defense but also contribute to biofilm formation, acting as a protective shield against antibiotic penetration. 9
Taking advantage of ongoing MDR surveillance and a well-established biobank of MDR bacteria, iGC initiated the iPHaGe (icddr,b Phage Hunting and Genomics) project. This initiative aims to uncover therapeutic phages and establish a dedicated phage biobank for potential medical applications. Preliminary findings identified 83 phages from 11 hospital effluent samples against 13 different Enterobacteriaceae host strains. Two notable phages, iPHaGe-KPN-11i and iPHaGe-KPN-12i, showed promise against extended-spectrum cephalosporin-resistant (ESCr) K. pneumoniae and other Enterobacteriaceae as well as secondary hosts; details of which are elucidated in this article.
Materials and Methods
Bacterial strains and culture conditions
An ESCr K. pneumoniae isolate ARCH-BD-1468 (NCBI SRA accession no. SRR26688493) was used as the host bacterium for the initial bacteriophage hunting under the iPHaGe study. The host species sequences type (ST152) and drug-resistance profile were confirmed by whole genome sequencing (WGS). An antibiotic sensitivity test was also performed to correlate the genetic drug-resistant profile with the phenotype profile for this clinical isolate (Supplementary File S1). The bacterial host was retrieved from the iGC archive (stored at −80° in 15% v/v-glycerol) and cultured in Luria–Bertani broth at 37°C for >16 h with shaking at 150 rpm.
Isolation, propagation, and purification of phage
Hospital effluents from Dhaka city were collected in a clean sterile bottle and transferred to the iGC laboratory for coculture with the host within 3 h of collection time. Freshly collected 4.5 mL of hospital effluents was mixed with 0.5 mL 10 × Luria–Bertani broth and 1 mL overnight host culture, then incubated at 37°C for >16 h with shaking at 150 rpm. After coculture, samples were centrifuged at 8000 g for 10 min and passed through 0.22 μm pore-size membrane to prepare the lysate.
Direct spot test (DST) was used to confirm the lytic phage, and the double-layer agar (DLA) method was used to isolate a single plaque for further phage purification and propagation. A single plaque was inoculated in 3 mL Luria–Bertani broth with 200 μL host and cultured overnight. The phage lysate was again collected from purified plaque culture and confirmed the presence of lytic phage by DST as well as uniform plaque morphology by DLA assay. This lysate was used for further experiments.
In vitro characterization of purified phage
Host range
Broad spectrum host range was observed by DST using an additional 25 bacterial strains with different resistance patterns (Supplementary File S1).
Optimal multiplicity of infection
Multiplicity of infection (MOI) for phages were determined by growing serial dilutions of K. pneumoniae isolate ARCH-BD-1468 until it reached its early exponential phase using 96-well microtiter plate assay. 10 The stock solution of bacteria (1 mL) and phages were added at varying MOIs (0.0001–10,000). The desired MOIs were achieved by fine-tuning the concentrations of both bacteria and phage, resulting in a final volume of 300 μL per well in the microtiter plate.
Each MOI was tested across five replicate wells. Control measures included media alone (absent of bacteria and phage), phage alone (absent of bacteria), and bacteria alone (absent of phage). The absorbance values at OD-600 for each well were documented at 30 min intervals over 6 h using a microplate reader (Dynex MRXe, Germany). The average absorbance readings for each MOI and controls were then graphed over time to analyze the MOI's impact on bacterial growth.
One-step growth curve
A one-step growth curve was calculated to determine the latent period 11 by monitoring the fluctuating count of phage particles during a replication cycle. In brief, the host strain ARCH-BD-1468 was cultivated to its exponential phase at 37°C and combined with isolated phages at an MOI of 0.1. At 5-min intervals, samples from the infected culture were serially diluted to enumerate the phages using DLA. This procedure was carried out in triplicate.
Statistical analysis
Every experiment was performed at least three times, with the outcomes presented as mean ± standard deviation. Graphs and statistical evaluations were conducted using the R program.
Phage DNA extraction
PEG-8000 concentrator was prepared by dissolving 80 g PEG-8000, 14.0 g NaCl and 20 mL of 10 × phosphate buffered saline (pH 7.4) in 80 mL Milli-Q water, then adjusted to 7.0–7.2 pH and 200 mL final volume. Three-milliliters of concentrator solution was mixed with 12 mL of phage lysate (>1 × 1012 PFU/mL) and incubated overnight at 4°C. After incubation, the mixture was centrifuged at 20,000 g for 15 min, and the pellet was resuspended with 5 mM MgSO4. To degrade and precipitate the proteins, 100 μL lysis solution (20 mM tris-HCl, 150 mM KCl, and pH 7.5) and 25 μL of Proteinase K (>600 mAU/mL) were mixed with 500 μL concentrated sample and incubated for 1 h at 56°C.
An equal volume of phenol:chloroform:isoamyl alcohol (25:24:1) was integrated, and 500 μL supernatant was transferred after centrifugation at 14,000 g for 10 min at 4°C. After centrifugation, an equal volume of isopropanol and 100 μL sodium acetate were added to the supernatant and kept for 30 min at −20°C. The samples were then centrifuged again, and the final wash was accomplished by adding 70% ethanol and letting it sit for 1 min at room temperature. Finally, pellets were air dried and DNA was dissolved in 100 μL of TE solution and stored at −20°C. DNA concentration was confirmed by NanoDrop and Qubit (ThermoFisher).
Sequencing and in silico analysis
The sequence library was prepared using the NEBNext Ultra II FS DNA Library Prep Kit, sequenced in Illumina MiSeq and subsequently analyzed; detailed methods were provided in our previous article. 12 The paired-end raw reads (233,803 for iPHaGe-KPN-11i and 292,080 for iPHaGe-KPN-12i) were checked for quality and after subsequent analysis assembled to generate the contigs. The complete genome was annotated using the online server RAST (https://rast.nmpdr.org/), with the annotation verified with pharokka v1.5.1 (https://github.com/gbouras13/pharokka) 13 and visualized using the Proksee online server (https://proksee.ca/). The NCBI webserver local alignment search tool BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was employed to confirm protein functions.
For the comparative analysis of two sequenced phages, the R package gggenomes (https://github.com/thackl/gggenomes) was utilized.
Results
Two out of six hospital effluents were DST-positive against the primary host (ESCr K. pneumoniae isolate ARCH-BD-1468). Overnight coculture of the hospital effluents yielded 2.9 × 107 and 3.0 × 107 PFU/mL with an average plaque diameter of 0.8 ± 0.2 mm. The plaques morphology (clear, round, and non-halo plaques) was similar for both positive hospital effluents (Fig. 1A). A single PFU from each positive hospital effluent was purified and named as iPHaGe-KPN-11i and iPHaGe-KPN-12i.
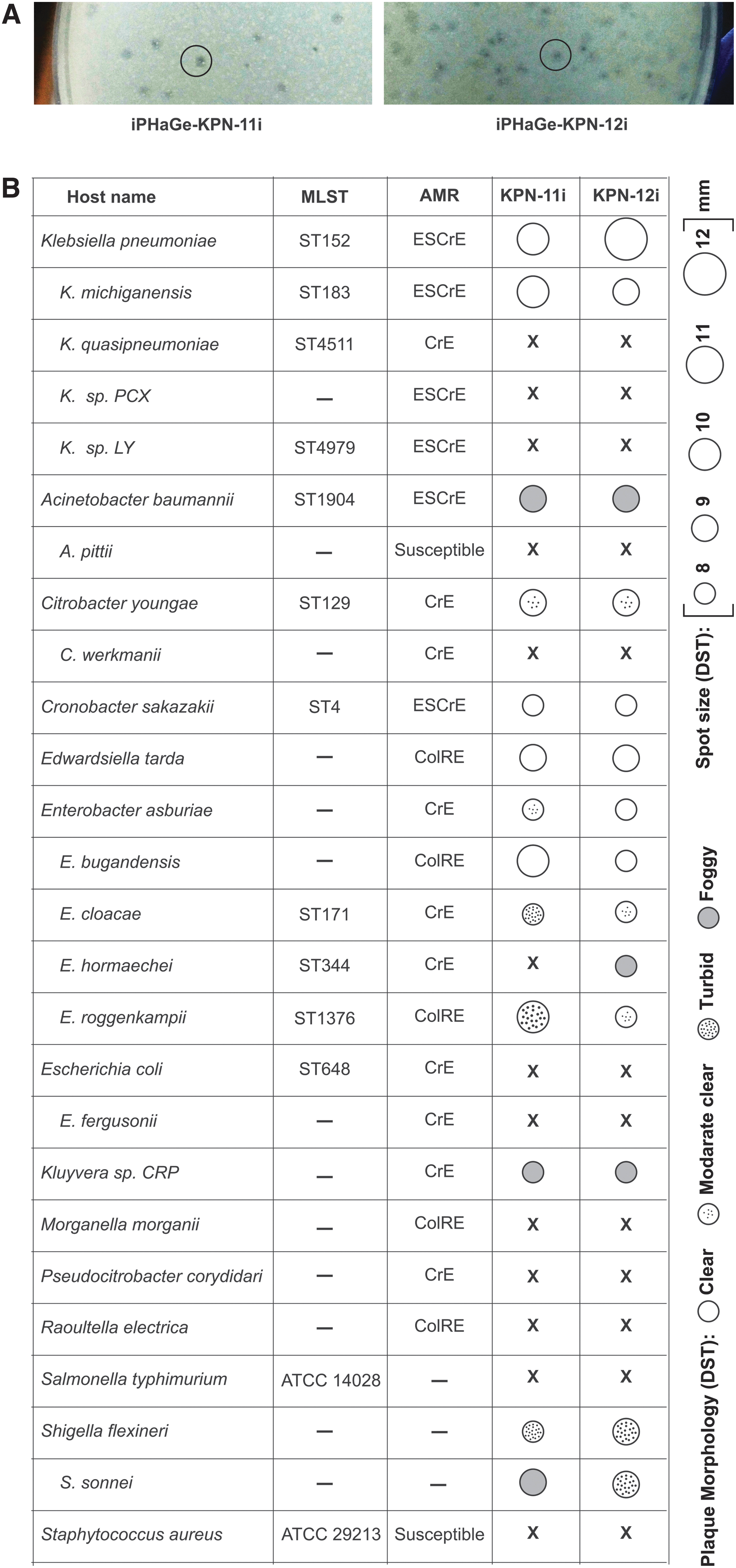
Bacteriophage host range and relative efficiency of plating
The DST assays revealed variations in host ranges for the two phages, as indicated by spot size and morphology differences. The phage iPHaGe-KPN-11i demonstrated the ability to lyse 12/26 strains, with 4 displaying clear lysis zones. In contrast, phage iPHaGe-KPN-12i exhibited lytic activity against 13/26 strains, with 5 showing clear lysis zones (Fig. 1B).
MOI and one-step growth curve
Bacteria subjected to higher MOIs demonstrated a rapid decline in OD values. By the 6 h mark, the untreated bacteria displayed the highest peak of OD (1.3 ± 0.02) (Fig. 2A). For iPHaGe-KPN-11i, bacterial growth curves with MOI <0.001 showed similar results to the untreated control, and MOI 1 showed a stationary phase after 2 h of incubation. Although iPHaGe-KPN-12i showed lower MOI, even with MOI 0.0001 the OD was lower than untreated bacterial control (Supplementary File S1) and with MOI 0.001, the host went to the stationary phase after 2 h of incubation.
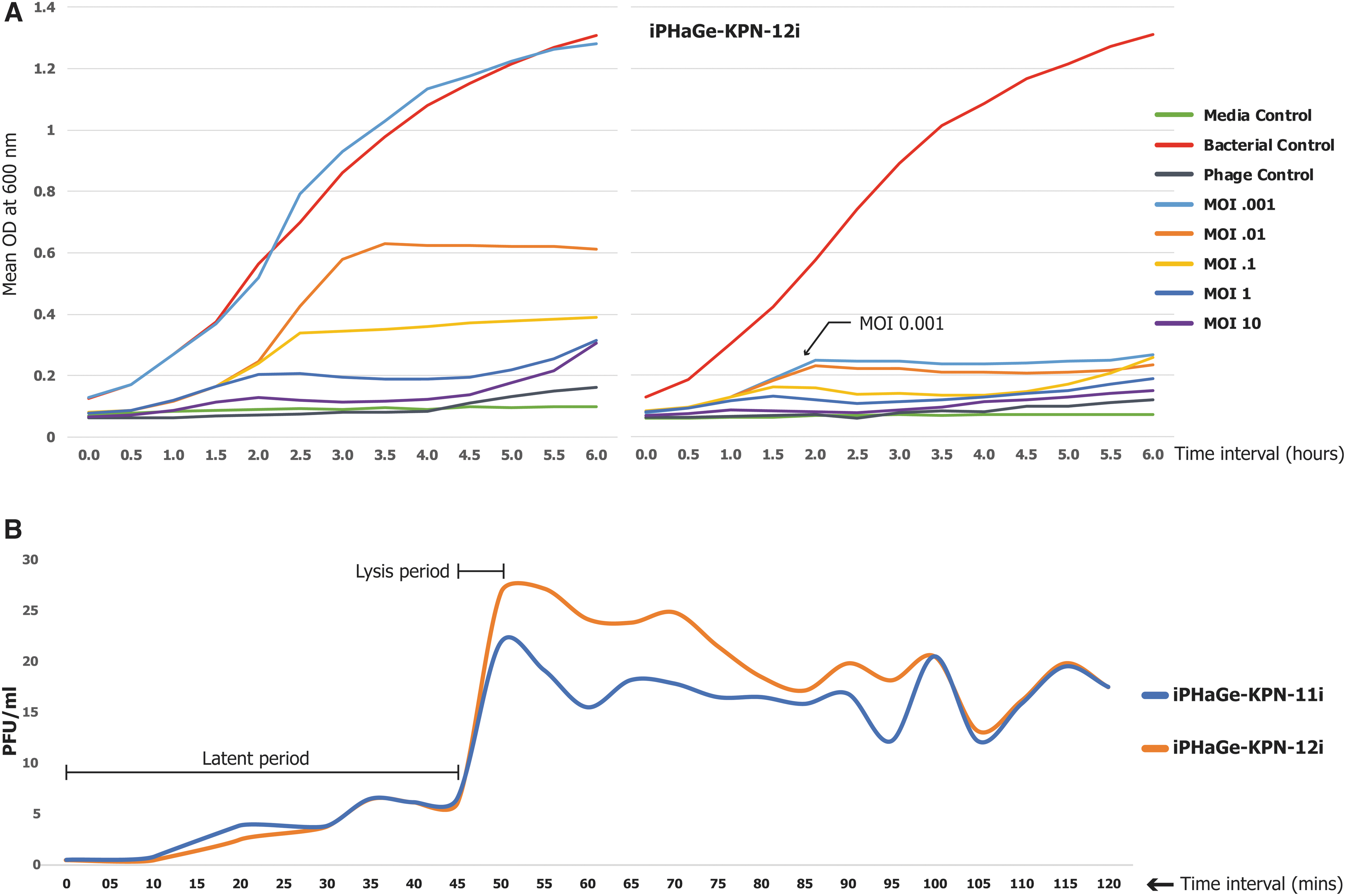
The observed phage replication kinetics in the one-step growth experiment revealed that both phages had about 45 ± 2 min latent periods, followed by <5 min lysis (Fig. 2B).
Genome annotation and bioinformatics analysis
WGS of phages indicated that both belong to the Pseudotevenvirus genus of Straboviridae family but vary in genome size and the number of encoded proteins (Table 1). The NCBI BLASTn identified the closest genomes of the phages previously identified against Klebsiella, Cronobacter, and Enterobacter.
Genome Characteristics and Comparative Analysis of iPHaGe-KPN-11i and iPHaGe-KPN-12i with the Closest Relatives
In brief, iPHaGe-KPN-11i has a 177,603 bp (GC 44.9%) genome with 273 coding sequences (CDS), whereas iPHaGe-KPN-12i has a slightly larger genome of 178,179 bp (GC 44.7%), with 275 CDS. At least 120 putative proteins have been identified with functions ranging from DNA replication/transcription/repair to DNA packaging, structure formation, and cell lysis.
The annotation map was constructed using the iPHaGE-KPN-12i genome structure then the iPHaGe-KPN-11i genome was super-imposed with it to identify similar coding regions (Fig. 3A). However, the regions of gene similarities indicated that the same gene did not share same position through the genome of these two phages (Fig. 3B). Further genomic assessments revealed that both phages encode two tRNAs and a single endolysin gene, phage lysozyme (PF00959.22).
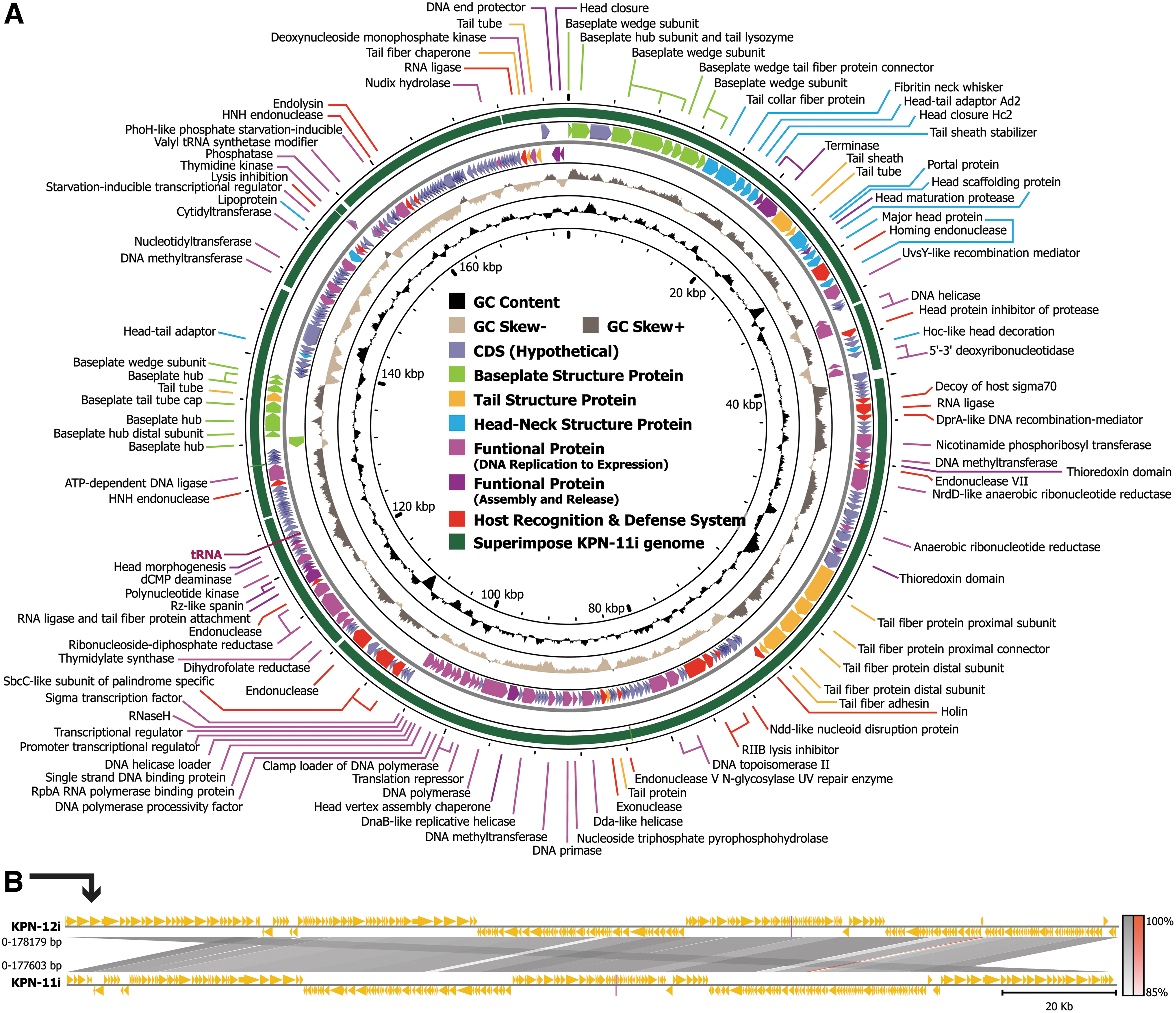
In addition, both phages had five protein-coding genes associated with endonuclease activity (Homing endonuclease, HNH endonuclease, Putative-endonuclease, Endonuclease-V, and Recombination-endonuclease-VII), three linked to exonuclease activity [3′-5′ exonuclease (dexA), exonuclease-subunit-1, and exonuclease-subunit-2], and four designed to counter host defense mechanisms (Holing, T4-RNA-ligase-1, RNA-ligase-2, and DNA-[N6-adenine]-methyltransferase [Dam]). None of the identified open reading frames (ORFs) suggest the presence of lysogenic phage-related proteins, such as transposases or integrases. Moreover, when the phage genome was analyzed using PhageLeads, 14 no genes associated with temperate life cycles, antibiotic resistance, or bacterial virulence were detected.
Discussion
The rise of K. pneumoniae resistant to various antibiotics is a significant concern for global health. Given the diminished efficacy of antibiotics, the potential of phages to eliminate recalcitrant K. pneumoniae infections is increasingly evident.15–17 This research highlights two promising phages, iPHaGe-KPN-11i and iPHaGe-KPN-12i, collected from hospital effluents in the highly populated country, Bangladesh. In this study, we detail their extended host range, MOI, One-Step Growth Curve, and genomic characteristics.
To be considered for therapy, comprehensive characterization of a phage is essential. Both of our phages exhibited promising outcomes after characterization; however, in terms of host range and MOI, iPHaGe-KPN-12i was better than iPHaGe-KPN-11i. Recent advancement in next-generation sequencing technologies has opened up exciting possibilities for WGS of phages that allow quick identification and characterization. In addition, it allows for investigating in silico potential host–pathogen interactions. The presence of genes linked to antibiotic resistance or bacterial virulence, which might be transferred from the phage to bacterial hosts, is considered undesirable when selecting phages for therapeutic purposes. 18
WGS data of our phages revealed an absence of genes linked to virulence and/or antibiotic resistance, emphasizing the potential safety and suitability of these phages for therapeutic applications. In addition, a phage's ability to infect various hosts is a crucial aspect, often related to the expression of multiple carbohydrate depolymerases in their tail fibers. 19 In our study, the presence of various ORFs associated with tail-fiber proteins and DST results underscored the broad lytic spectrum of these phages. Furthermore, the potential host lysis protein genes identified during the in silico analysis offer promising avenues for the development of future therapeutics.
Using a single MLST of K. pneumoniae (ST152) strain to observe lytic capability may not be ideal and could be considered as a study limitation for the host range analysis. Nonetheless, we plan to utilize the ongoing MDR surveillance of iGC and its comprehensive biobank containing 156 MLSTs of K. pneumoniae to validate the host range and provide a comprehensive picture. Another constraint of this research was the omission of phage morphology through electron microscopic examination. However, genomic data were employed to pinpoint closely related phages with verified microscopic structures, which might be consistent with our studied phages.
Conclusion
Unlike many host-specific phages, these two demonstrated a broader lytic spectrum; enabling the opportunity to use them in phage cocktails for host range expansion with further evaluations and could be a beacon of hope in the looming darkness of the AMR crisis.
Data Availability
WGS data of the phages iPHaGe-KPN-11i and iPHaGe-KPN-12i are available in the NCBI's Sequence Read Archive under accession numbers SRX20726928 and SRX20726929; the annotated genome assembly is available in NCBI GenBank under accession numbers OR637327 and OR437326, respectively.
Footnotes
Acknowledgments
We would like to express our sincere gratitude to Dr. Zachary Ardern, postdoctoral scholar in the Thomson Group at the Wellcome Sanger Institute, United Kingdom, for his invaluable contributions and insightful comments during the review of our article. We also gratefully acknowledge icddr,b core donors (the governments of Bangladesh and Canada) for their unrestricted support and commitment to icddr,b's research efforts.
Authors' Contributions
M.R. conceived the study and provided all support to conduct the laboratory investigation. S.R. optimized laboratory protocol, coordinated laboratory investigation, analyzed data, interpreted the results, and wrote the article. J.A., I.M., A.H., and S.I. performed laboratory procedures. M.M.H. and T.K. analyzed and interpreted genomic data. M.J. coordinated all laboratory procedures. S.I.M., A.B., S.K.B., M.J., and M.R. critically reviewed the article and provided intellectual input. All authors reviewed subsequent drafts of the article, approved the final version, had full access to all the data in the study, and accepted the responsibility to submit for publication.
Author Disclosure Statement
No competing interests exist.
Funding Information
The study was supported by icddr,b core funds.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.