
Abstract
Alzheimer disease (AD) is associated with mitochondrial dysfunction. In this study, we investigated succinic dehydrogenase (SDH) activity in mitochondria of hippocampal CA1 pyramidal neurons obtained from 10-month-old 3xTg-AD mice, an animal model of AD, as well as from age-matched control mice PS1-KI. In SDH-positive mitochondria, we measured numeric density (Nv, number of mitochondria/μm3 of cytoplasm), average organelle volume (V), volume density (Vv, volume fraction of mitochondria/μm3 of cytoplasm), average length (Fmax), and the ratio (R) between the total area of the cytochemical precipitate due to SDH activity and the total mitochondrial area. Our results indicate that 3xTg-AD mitochondria show a significant decrease of Nv, increase in V and Fmax, as well as a trend toward a reduction of R, whereas Vv is unchanged. Our findings further support the idea that mitochondrial dysfunction is involved in AD and are in line with studies indicating that both amyloid precursor protein (APP) and amyloid-β (Aβ) localize to mitochondria.
Introduction
Succinic dehydrogenase (SDH) is the key component of complex II of the oxidative phosphorylation reaction (OXPHOS) as well as a secondary electron entry point of the electron transport chain. At variance with other OXPHOS molecules, SDH is completely encoded by the nuclear genome and is then transported into the mitochondrion to be included in the inner mitochondrial membrane. SDH is the only mitochondrial enzyme that is used for both electron and carbon fluxes, thereby representing a unique molecule for a critical cross control between cellular respiration and the Krebs cycle. 6,7 On the basis of these features, a quantitative evaluation of SDH activity may provide reliable information on mitochondrial dysfunction and an indirect index of how adequate the adenosine triphosphate (ATP) supply is. 8 –10
A triple-transgenic model (3xTg-AD) for AD has been recently developed and widely employed to better understand the molecular mechanisms underlying the AD-related pathology. 11 These triple-transgenic mice overexpress mutant amyloid precursor protein (APP), presenilin 1 (PS1), and tau and develop both plaque and tangle pathology in discrete brain regions. Notably, 3xTG-AD mice show functional deficits in synaptic plasticity, including long-term potentiation (LTP) alterations, that occur before extracellular amyloid-β (Aβ) deposition and tangles formation, but are instead strongly associated with the appearance of intraneuronal Aβ deposition first in the neocortex and then in CA1 pyramidal neurons. 11 In support of these findings, we have found that 13-month-old 3xTG-AD mice show a decrease in the fraction of perforated synapses (−28.5%), 12 suggesting that alterations in the enlargement–perforation–splitting cycle at CA1 junctional sites may occur with consequent impairment of synaptic plasticity. In the present study, we investigated SDH-positive mitochondria of 3xTG-AD mice with the intent of exploring early signs of metabolic dysfunction involved in AD-related pathology.
Material and Methods
Four 3xTg-AD and four age-matched control (PS1-KI) mice (10 months of age) were used for the present study. Transgenic animals were from colonies kept at CeSI (Center of Excellence on Aging, University “G. D'Annunzio,” Chieti, Italy) and originated from breeding pairs kindly obtained from Frank LaFerla. The animals were treated in accordance with the European Community guidelines on animal care. Mice were anesthetized with an intraperitoneal injection of 2,2,2-tribromoethanol (200 mg/kg body weight) and sacrificed. Brains were rapidly removed, and hippocampi were excised and immediately sectioned into thin slices.
Samples were investigated for evidence of SDH activity by employing the copper ferrocyanide cytochemical reaction. 13 Briefly, 3xTg-AD and PS1-KI samples were immersed for 45 min at 37°C in either substrate or substrate-free incubation media (see refs. 9, 10, and 14 for a detailed description of the media). Postfixation, dehydration, and embedding were then carried out with conventional electron microscopy procedures. SDH-positive mitochondria were stained darkly against a faint background. The rationale of this method relies on the principle that the amount of cytochemical precipitates correlates with SDH activity and provides a reliable estimation of the metabolic competence of any individual mitochondrion. Electron microscopy pictures of SDH-positive mitochondria were taken with a digital camera directly connected to our image analysis system (Kontron KS300, Munich, Germany).
The sampling of SDH-positive mitochondria was carried out in the perikaryon of CA1 hippocampal pyramidal neurons in accordance with the “equal opportunity rule.” 15 For each animal, 500 μm2 of perikaryon was analyzed, yielding a total sampled area of 2000.00 μm2 for each experimental group (i.e., 500 μm2 × 4 3xTG-AD or PS1-KI mice). The following parameters were analyzed by applying previously employed morphometric formulas 16 : numeric density (Nv, number of mitochondria/μm3 of cytoplasm), average volume (V), volume density (Vv, overall volume of mitochondria/μm3 of cytoplasm), and average length (Fmax) of SDH-positive mitochondria. The ratio (R) between the overall area of the cytochemical deposits due to the SDH activity and the area of the SDH-positive mitochondria was also calculated and expressed as fraction of mitochondrial membrane area involved in SDH activity.
Statistical analysis was performed by employing the Student t-test.
Results
The staining procedure employed to detect SDH activity is based on the reduction of ferricyanide to ferrocyanide that is related to the activity of the enzyme. The reaction develops an electron-opaque and insoluble copper ferrocyanide precipitate that is trapped at the mitochondrial membrane. SDH-positive mitochondria of 3xTg-AD mice show a statistically significant decrease of Nv (Fig. 1A), a parallel significant increase of V (Fig. 1B) and Fmax (Fig.1C), while Vv remains constant (Fig. 1D). Moreover, in 3xTg-AD mice, more than 36% of SDH-positive mitochondria show an average volume greater than 0.25 μm3, whereas in control PS1-KI mice only 11% of the organelles are of this size (Fig. 1E).
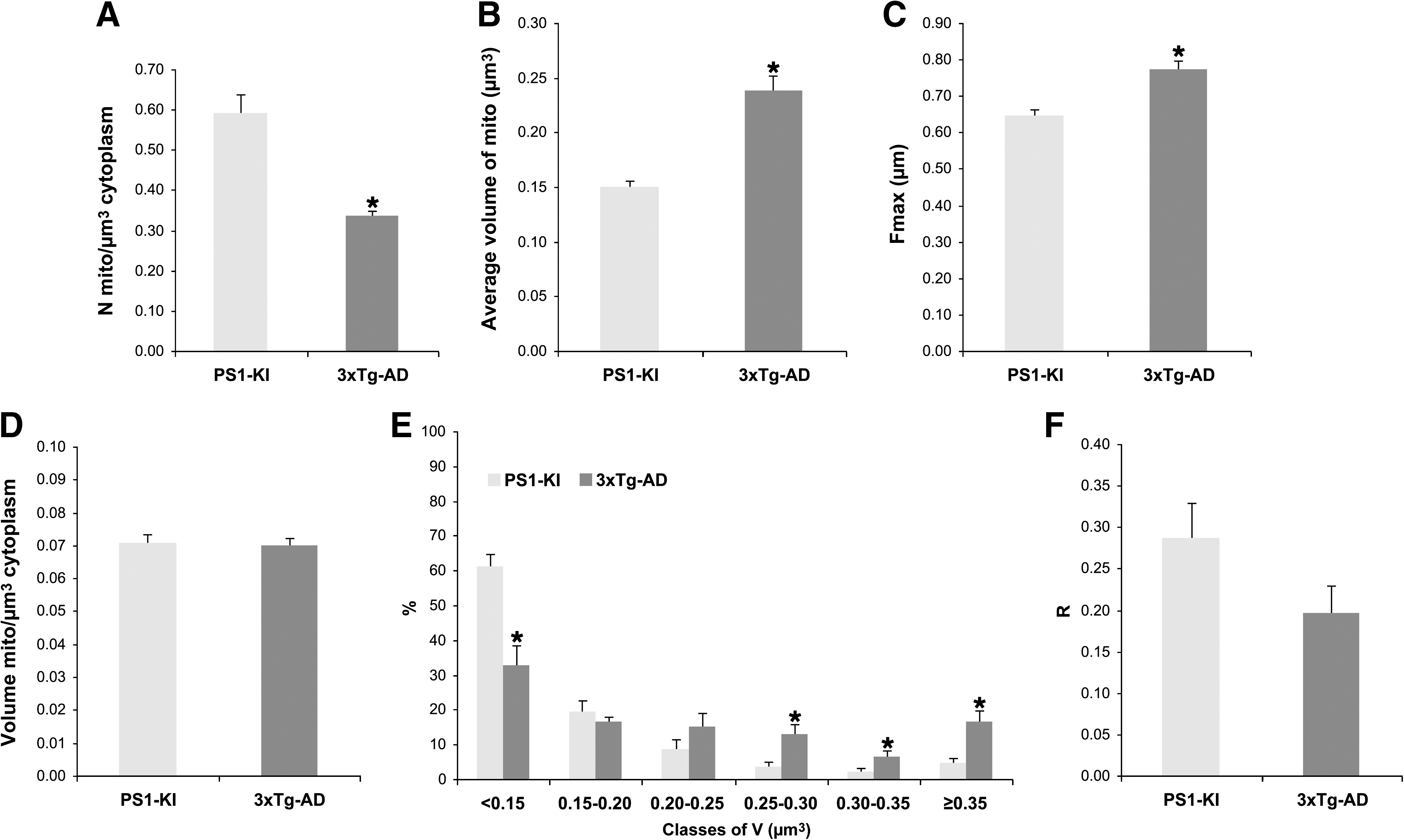
Ultrastructural parameters of succinic dehydrogenase (SDH)-positive mitochondria from the perikaryon of CA1 hippocampal pyramidal neurons. (
When analyzing the R value, we found a trend toward reduction (by 31%; Fig. 1F) in 3xTg-AD mice when compared to PS1-KI mice; however this reduction did not reach statistical significance.
Discussion
In this study, we found a selective decrease in the numeric density of SDH-positive mitochondria in the perikaryon of CA1 hippocampal pyramidal neurons from 3xTg-AD mice. Moreover, the morphometric analysis revealed that SDH-positive mitochondria increase their size (V, +58%), a phenomenon that is likely due to a marked elongation of their shape (Fmax, +20%). Mitochondrial division in SDH-positive organelles appears to be impaired, but the higher values of V and Fmax support the concept that 3xTg-AD mice compensate for the reduction in Nv, and the final outcome is an unchanged value of volume density (Vv).
It should also be emphasized that succinate (as well as glutamate-malate) supports maximum rates of respiration, therefore quantitative cytochemistry of this molecule can be taken as a reliable index of neuronal metabolic competence when energy demand is high. 7,8 With specific reference to neurons, cells that are characterized by frequent bursts of activity, this feature represents an almost physiological condition that has to be met to supply adequate amounts of ATP. Thus, the mitochondrial capacity to provide ATP in neurons is continuously challenged by their physiological high levels of performance, and the estimation of SDH activity may constitute a reliable and sensitive metabolic marker that is likely to be proportional to energy production needs.
The causative events leading to the impairment of SDH-positive mitochondria and the decrease of R values in the 3xTG-AD mice are not known, but increased production of free radicals, defects in the mitochondrial genome, derangements in the interaction between nuclear and mitochondrial DNA, damage of the mitochondrial membrane, as well as changes in its lipid composition are among the factors that can be hypothesized to play a synergistic role. Aβ has been shown to directly inhibit mitochondrial energy metabolism in neurons, causing reduced ATP levels, decreased activities of the respiratory chain complex, depolarization of the mitochondrial membrane, decreased oxygen consumption, 17 and, ultimately, neuronal death.
Interestingly, brain tissue samples from AD patients show a significant decrease in the number of intact mitochondria and a loss of mitochondrial enzymatic activity that is associated with an increase in lipofuscin deposits. 18 Intracellular Aβ aggregates have been observed and interpreted as an early sign of AD-related pathology in APP-overexpressing mice, and it has been suggested that APP and Aβ might act intracellularly to promote mitochondrial dysfunction. 19 Furthermore, immunoelectron microscopy studies have shown that, in a transgenic AD mouse model, 20 APP targets mitochondria of selected brain regions and is in stable contact with two proteins, TOM40 and TIM23, involved in the mitochondrial import machinery, thereby blocking the import of mitochondrial preproteins. Studies on post mortem brain samples from AD patients and control subjects have shown that APP is present in mitochondria of AD patients but not in the organelles of control patients.
Altogether, these findings support the hypothesis that APP interferes with mitochondrial respiration and causes decreased importation of respiratory chain subunits, increased hydrogen peroxide (H2O2) production, impaired mitochondrial reducing capacity, as well as defective brain energy metabolism. Moreover, other studies have documented that also Aβ interacts with mitochondria and impairs their functions as well as increases free-radical generation. 21 In conclusion, our findings in 3xTg-AD mice support the idea that an impaired SDH activity can play a role in the early steps of the AD-related neurodegenerative cascade.
Footnotes
Acknowledgments
The authors sincerely thank Mr. Moreno Solazzi for his skillful technical help and Mrs. Lucia Montemurro for the graphic support. The study was partially supported by ISS (Istituto Superiore di Sanità) grant no. 533F/E/1.