
Abstract
We demonstrate that intravenous delivery of human, or rat, pancreas-derived pathfinder (PDP) cells can totally regenerate critically damaged adult tissue and restore normal function across a species barrier. We have used a mouse model of streptozotocin (STZ)-induced diabetes to demonstrate this. Normoglycemia was restored and maintained for up to 89 days following the induction of diabetes and subsequent intravenous delivery of PDP cells. Normal pancreatic histology also appeared to be restored, and treated diabetic animals gained body weight. Regenerated tissue was primarily of host origin, with few rat or human cells detectable by fluorescent in situ hybridization (FISH). Crucially, the insulin produced by these animals was overwhelmingly murine in origin and was both types I and II, indicative of a process of developmental recapitulation. These results demonstrate the feasibility of using intravenous administration of adult cells to regenerate damaged tissue. Critically, they enhance our understanding of the mechanisms relating to such repair and suggest a means for novel therapeutic intervention in loss of tissue and organ function with age.
Introduction
The exact origins and nature of cells required in vivo to repair pancreatic damage remains a complex and controversial topic. In the developing embryonic pancreas, β-cells arise from endocrine progenitor cells expressing the transcription factor neurogenin 3 (Ngn3). 1,2 However, this progenitor population is ephemeral and, postnatally, β-cell maintenance and neogenesis derive from the slow proliferation of preexisting, terminally differentiated, β-cells. 3 –7 A further mode of β-cell replacement has been reported in adult mice following pancreatic injury by partial duct ligation, involving Ngn3 positive facultative endocrine progenitors in the ductal lining. 8 This latter observation is intriguing, because it indicates that the adult pancreas has the capacity to re-engage an embryonic mode of β-cell generation. The molecular mechanisms of these observed regeneration events are unknown.
Previously, pancreatic ductal epithelial cells with the potential to dedifferentiate into a progenitor cell capable of proliferating and forming new islets and acini have been described. 9 As ductal cells replicate, they transiently express PDX-1, a transcription factor that is expressed in embryonic pancreatic precursors. This has been hypothesized to be a less restricted phenotype that can serve as a multipotent progenitor. 9 However, definitive demonstration is awaited. Recently, CK19-expressing nonendocrine pancreatic epithelial cells (NEPCs) were reported to be partially induced to differentiate into insulin-producing cells in vivo, in the presence of fetal pancreatic tissue, but no evidence for β-cell replication in this model was demonstrated. 10
Recently, we isolated and characterized a novel cell population from adult rat and human pancreas termed pancreas-derived pathfinder (PDP) cells. 11 As with previously described progenitor/stem cells from the pancreas, 10,12 this cell population can be induced to form islet-like structures in vitro when cultured in nicotinamide-containing differentiation media. However, we have also described a subpopulation expressing PDX-1, but not CD90, that failed to demonstrate morphological changes under the same in vitro conditions, yet produced insulin transcripts. 11
We sought to determine how PDP cells could contribute to islet formation in vivo in the presence of pancreatic tissue, with a view to determining their potential as a direct cellular therapy for diabetes, or as mediators of pancreatic tissue regeneration. For these experiments, we investigated the utility of direct intravenous delivery of rat and human PDP cells after diabetes induction in mice.
To track the transplanted cells, we used a xenogeneic model in which male Albino Swiss (AS) rat or human PDP cells were given to female C57BL/6 mice, rendered diabetic by high-dose STZ, a well-defined model of type 1 diabetes. 13,14 This allowed us to address whether the PDP cells differentiated in vivo to repair damage, stimulated the host tissue to repair damage, or both.
Materials and Methods
Isolation and maintenance of PDP cells
Pancreatic ductal tissue was isolated from 12-month-old Albino Swiss (Glasgow) rats, and an undiseased female human pancreatic biopsy. In both instances, tissue was microdissected and minced, prior to seeding in CMRL medium (Invitrogen, Paisley, UK). The PDP cells emerged as a confluent monolayer after approximately 5 weeks in culture. These were then harvested and washed in phosphate-buffered saline (PBS). PDP cells were maintained in culture in 20 mL of CMRL 1066 medium supplemented with 10% fetal bovine serum (FBS; Sigma, Poole, UK), 2 mM glutamate, 1.25 mg/mL amphotericin B, and 100 U/mL penicillin, 100 μg/mL streptomycin (all Invitrogen, Paisley, UK) in T75 culture flasks with 0.2-μm filter caps (Corning, UK) at 37°C in a 5% CO2 atmosphere.
STZ treatment and PDP cell transplantation
Female C57Bl/6 mice (n = 4) were made diabetic by injection of STZ to give a dose of 250 mg/kg on day 0 of the experiment. Male rat or female human PDP cells were injected into the tail vein on day 3 after STZ injection. STZ was prepared in 20 mM citrate buffer (pH 4.5) and used immediately for intraperitoneal delivery. Control animals were given an injection of conditioned culture medium. Blood glucose was monitored every 3 days. All procedures were conducted under the authority of the UK Home Office (HO) and performed as described previously. 15
Under these licensing conditions, animals with blood glucose above 30 mM/L or a body weight loss of 20% or more were required to be sacrificed. In this protocol, diabetes is defined as serial nonfasting glucose levels, 3 days post-STZ and beyond, of >20 mM/L. Furthermore, severe progressive hyperglycemia under the protocol used with these HO licensing conditions is defined as progressive rises in serum glucose to ≥30 mM/L or clinical signs of hyperglycemia.
Blood glucose measurement
Blood glucose values were determined following exsanguination via the tail vein, and measured using the Ascensia® AutodiscTM Blood Glucose Test Sensors (Bayer Healthcare, Berks, UK) following the manufacturer's recommendations.
Immunohistochemistry
Pancreata from PDP cell transplant and control mice were removed and snap-frozen in liquid nitrogen, then embedded in Tissuetek OCT compound (RA Lamb). Fixed frozen sections were rehydrated in PBS for 5 min. Endogenous peroxidase was quenched by incubating sections in 3% hydrogen peroxide (H2O2) for 10 min. Blocking was performed by incubating sections in casein solution (Vector) for 1 h at 25°C. The following antibodies were used as appropriate: C-peptide (human specific. Abcam, 1:100), glucagon (human specific, Serotec, 1:10; mouse specific, Cell Signalling Technology, 1:25; rat specific, Chemicon, 1:3,000), and insulin (mouse specific, Cell Signaling Technology, 1:100; rat specific, Affinity Bioreagents, 1:500). All antibody incubations were performed overnight at 4°C. In each run, negative (no antibody added to tissue) and positive controls were included. For these controls, normal untreated mouse pancreas was used. The purpose of the positive control was to demonstrate normal islet architecture and staining pattern for insulin, glucagon, and C-peptide. The negative control ensured that any staining detected was specific to each of the antibodies. Signal was visualized using a goat anti-rabbit secondary antibody (Dako, 1:200) for 1 h at 25°C followed by 3,3′-diaminobenzidine (DAB; Vector Laboratories).
Fluorescence in situ hybridization
Frozen sections were rehydrated in PBS, then pretreated with 1 M sodium thiocyanate at 80°C for 20 min. Tissue digestion was performed by treating sections with pepsin for 25 min at 37°C. Slides were then treated with 0.2% glycine for 5 min, then washed in Tris-buffered saline (TBS) and incubated with 4% paraformaldehyde for 5 min. Probes to detect rat Y (rat YCy3/12FITC, Cambio) and human X chromosomes (CEP X FITC probe, VYSIS) were used. The human X probe was prepared as per the manufacturer's instructions. Before use, both probes were denatured at 65°C for 10 min. The tissue was denatured in denaturation solution at 65°C for 1.5 min, then incubated in ice-cold ethanol for 2 min. Tissues were then dehydrated through a series of graded alcohols, before 10 μL of probe was added to each section. Probe and tissue were hybridized for 10 min at 80°C and then overnight at 42°C. Slides were then washed in posthybridization wash buffer for 5 min at room temperature, followed by a wash at 72°C for 2 min. Slides were allowed to air dry before being mounted onto coverslips using Vectashield (Vector).
Results
Full reversal of diabetes: Human or rat PDP cell treatment normalizes blood glucose
Under a HO-approved protocol allowing a 100-day follow-up, we injected 1.5 × 106 PDP cells into the tail vein of confirmed diabetic animals (n = 4 per group) on days 3 and 10 post STZ administration. A dose of 250 mg/kg STZ was used to ensure obliteration of β-cells, to the extent that untreated controls would not survive long enough to regenerate sufficient pancreatic tissue to mitigate the effects of the STZ treatment. Animals given STZ then became progressively hyperglycemic, such that they rapidly developed blood glucose levels above 15 mM/L, which then rose further with increasing time. Control animals were given equal volumes of conditioned medium. Blood glucose was monitored every 3 days for 4 weeks then weekly up until 89 days. PDP-cell-treated animals were sacrificed at 100 days for tissue analysis. Four treatment groups, of 4 animals each, were injected with either: (1) A mixed population of CD90+ and CD90− rat PDP cells; (2) CD90+ rat PDP cells; (3) CD90− rat PDP cells; or (4) a mixed population of CD90+ and CD90− human PDP cells. The subfractions of rat PDP cells, based on CD90 expression, were studied to determine if their contrasting in vitro differentiation capacities 11 correlated with their efficacy in vivo. No immunosuppression was administered with any treatment group, because preliminary experiments were consistent with the PDP cells being immunologically null. PDP cell administration in the presence of 20 mg/kg cyclosporin did not affect the capacity of PDP cells to reduce blood glucose levels (data not shown).
Immunohistochemical analysis indicated that 3 days after administration of STZ, pancreatic β-cells had been destroyed before the time point for PDP cell administration (Fig. 1B) in comparison to the untreated (no STZ) control (Fig. 1A). This dose of STZ is sufficient to render animals permanently diabetic for the time course of this experiment, because the conditioned medium–treated controls were profoundly diabetic 1 week after STZ administration and had to be sacrificed by day 12. Figure 1C records the last blood glucose measurement for these animals at day 9. All treated groups had significantly lower blood glucose levels at this time point (p < 0.01).
(
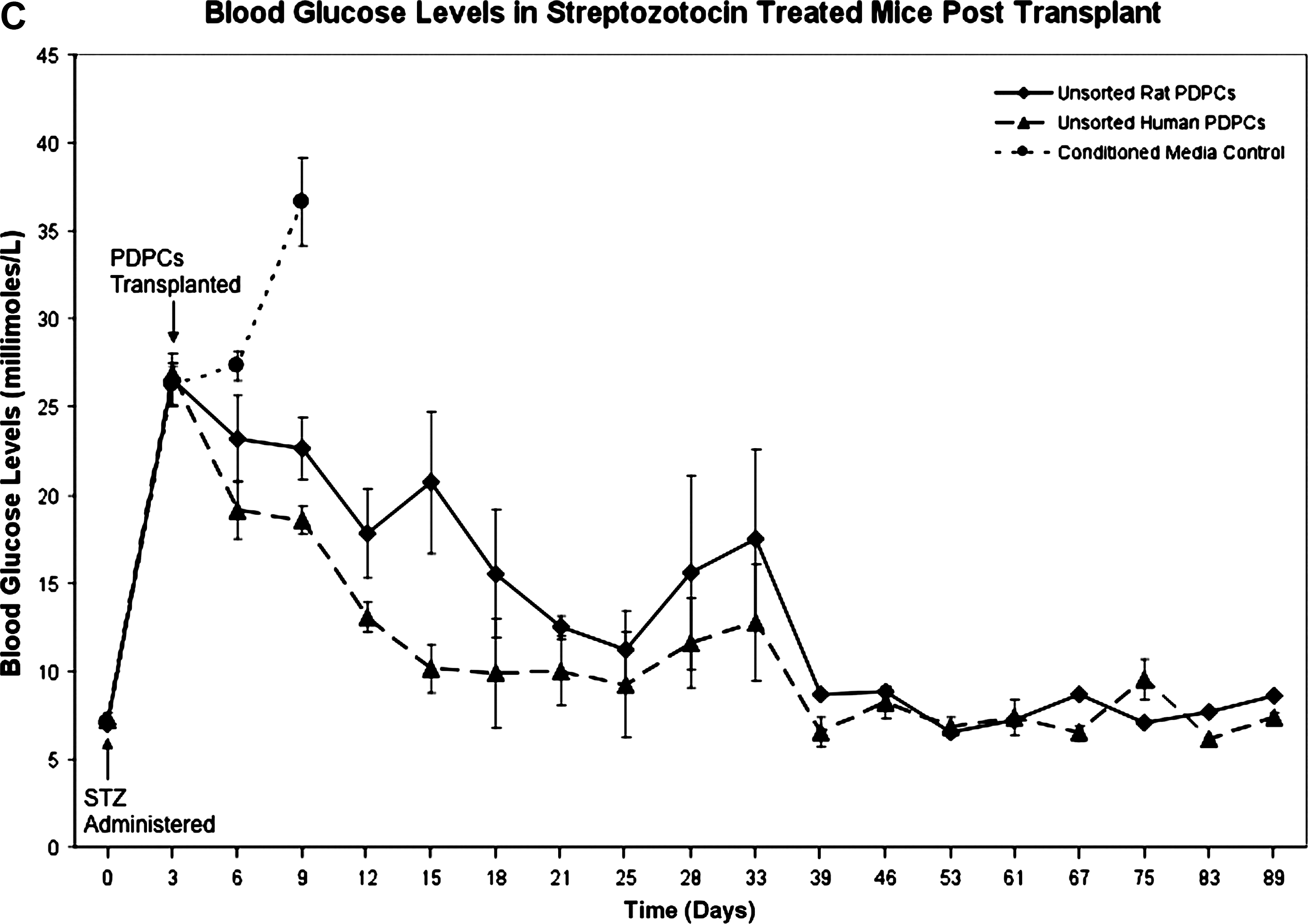
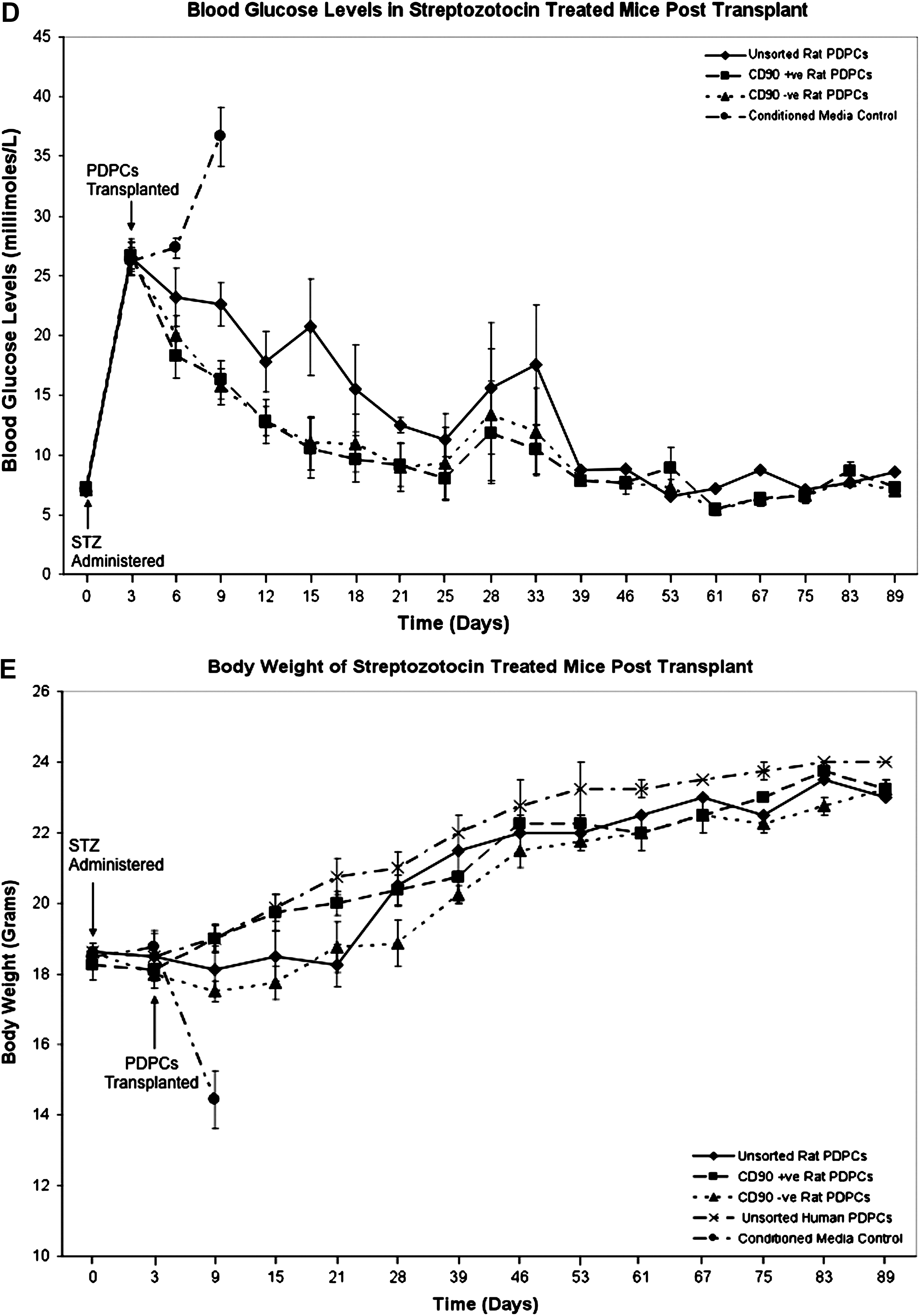
PDP cell-treated animals were still euglycemic 89 days post STZ administration when final blood glucose levels and body weights were recorded (Fig. 1C). In contrast, the control group, which developed progressive hyperglycemia, was sacrificed when blood glucose was >30 mM/L in keeping with HO-licensing conditions. These animals were profoundly diabetic at this time point, and their elevated blood glucose level, in contrast to PDP-treated groups, was not consistent with the occurrence of ongoing spontaneous pancreatic regeneration. No difference in efficacy was observed using CD90+ and CD90− PDP subfractions, despite the differences in the potentiality of these cells in vitro (Fig. 1D). 11 All PDP cell-treated mice showed a progressive reduction in blood glucose, which reached normal, pre-STZ treatment, levels by day 39 and maintained normoglycemia to the end of the experiment. This indicates that significant recovery of insulin production was achieved in these mice. Notably, no such reduction in blood glucose level was observed in controls, indicative that the dose of STZ used was sufficient to preclude survival due to pancreatic regeneration over the same time period. Animals treated with human or at CD90 subpopulations showed a consistent and greater initial reduction in blood glucose than the mixed rat CD90 group (Fig. 1 C,D), despite there being no significant difference in result (euglycemia) at day 89. The reason for this remains to be determined, but may simply reflect the small numbers of animals used, differences in cellular subfractions or batch variation, the result of minor operational errors amplified in vivo, undetermined host responses, or variability in animal feeding behavior.
Furthermore, the scenario of PDP cells enabling restoration of euglycemia is supported by the observation that all PDP cell-treated animals recovered from an initial loss of body mass and thereafter maintained a body weight consistent with age (Fig. 1E).
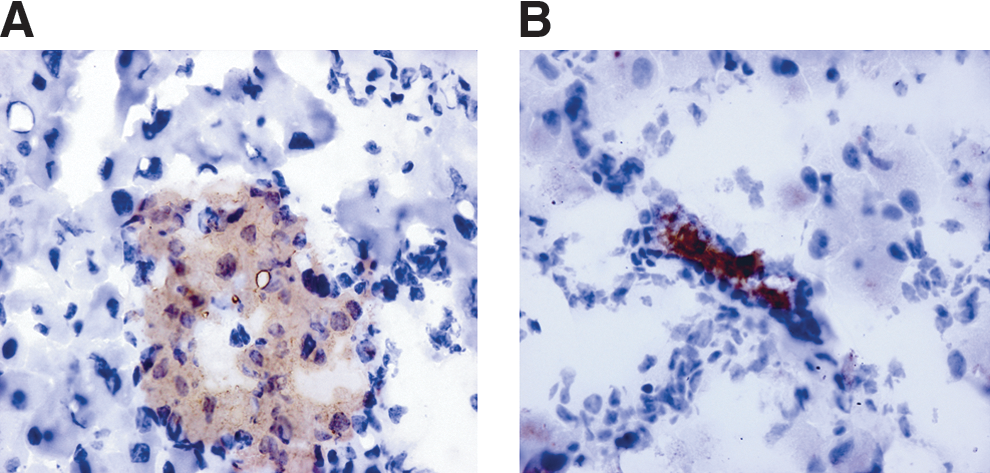
Consistent with the observed recovery of normoglycemia, immunohistochemisty indicated that PDP cell treatment was associated with regeneration of pancreatic β-cells and that the regenerated islets expressed insulin and glucagon, in a pattern that appeared normal for mice, with insulin staining the center of islets, whereas glucagon stained the outer shell (Fig. 2). C-peptide staining also appeared consistent with normal islet architecture (Fig. 2).

Streptozotocin (STZ) diabetic mice treated with rat and human pancreas-derived pathfinder cells (PDPCs) exhibit normal islet architechture at 100 days post PDP administration. Immunohistochemistry shown here demonstrates the presence and distribution of insulin, glucagon, and C-peptide cells within the islets of regenerated pancreas. STZ-treated mice were treated with either rat or human PDP cells. Brown staining represents insulin-, glucagon-, or C-peptide-positive cells in the respective panels. Individual panels are labeled to show the species of origin of the PDP cells. Normal untreated mouse pancreas was used as a positive control to illustrate the normal architecture and respective staining patterns for mouse pancreatic islets. The same tissue was used as a negative control (no antibody added) to ensure that any staining detected was antibody specific. Blue stain is Hematoxylin/Scotts Tap Water Substitute counterstain.
Detection of xenogeneic cells by fluorescence in situ hybridization (FISH) indicated that the overwhelming majority of the islet tissue was mouse in origin (Fig. 3). Only 0.05–0.1% rat signal was detected in the treated mice (n = 3), dependent upon the respective treatment group, with 0.18% being detected in the human cell–treated group (Fig. 3). Exhaustive examination of tissue sections (n = 30) revealed that no clusters of human or rat PDP cells were detected but instead, the xeno-signal was isolated and scattered throughout the sections, strongly suggesting that the injected PDP cells were not forming islet-like structures in vivo. Because we have no information about the occurrence of cell division, we do not know if these signals are from surviving original cells or from progeny of these cells. Therefore, we cannot make any statements concerning survival of the original cells.

Rat and human chromosomes are rarely detected by fluorescent in situ hybridization (FISH) in pancreas-derived pathfinder (PDP)-treated streptozotocin (STZ) diabetic mice. FISH detection for the rat Y chromosome and human X in the pancreas of STZ diabetic mice treated with PDP cells. Positive control hybridizations are shown to demonstrate the presence of the rat Y chromosome (red signal) and human X chromosome (green signal) in control rat tissue and human fibroblasts, respectively. These are labeled individually in the figure. Negative control hybridizations to demonstrate the absence of xenogeneic chromosome signal are similarly presented. Positive signal detection (red = rat Y; green = human X) for each respective species are highlighted by arrows in the lower four panels of the figure and similarly in the respective zoom panels. PDPC, Pancreas-derived pathfinder cells.
Detection of a scattered xeno-signal was also observed in the liver (0.05% rat and 0.07% human, respectively). The xeno-signal was observed in other tissues by polymerase chain reaction (PCR), including the brain, lung, heart, and kidney (data not shown). This signal, however, was not consistently detected in all tissues and in all animals tested, unlike within the pancreas, where there was consistent signal detection.
Reverse transcription (RT)-PCR analysis of pancreatic tissue in treated animals detected insulin transcripts that were both rat and mouse in origin. The RT-PCR-restriction fragment length polymorphism (RFLP) analysis indicated the presence of rat and mouse insulin gene (Ins II) transcripts. This was confirmed by cloning and sequencing of the relevant fragments. A RFLP analysis of the relative PCR amplicons from controls and treated animals is included in the Supplementary Data information for this manuscript. Significantly, PDP cell-treated mice also produced an Ins I transcript. This observation has been confirmed by the cloning and sequencing of the amplicon. Ins I is normally only produced during embryonic development in the mouse (please see Supplementary information; Supplementary Data are available online at
Although FISH analysis demonstrated that a small number of rat- or human-derived cells were present in treated animals, the numbers seen could not account for the nondiabetic phenotype observed. Extrapolating our cell count data suggests that xenogeneic cells could contribute in the range of 50 islet-equivalents (but not to actual islets) if all the cells observed had differentiated into insulin-producing cells. This maximum estimate would correspond approximately to an insulin production capacity of 5% of the total insulin made by the pancreas. This calculation is based on a mouse pancreas having approximately 1,000 islets, each with 1,000 β-cells. We detected a total of 595 xeno-signals in 495,500 nuclei. The latter corresponds to one twentieth of the number of β-cells in the pancreas (≈1,000,000) or ≈50 islet equivalents. This is sufficient to explain the observed rat Ins II mRNA. However, the great majority of insulin-producing cells in the regenerated pancreata derive from mouse host cells. Therefore, when taken altogether, these data are most consistent with PDP cell-mediated stimulation of endogenous pancreatic regeneration.
Discussion
Our data indicate that it is feasible to stimulate regeneration of critically damaged adult tissue, in this instance, pancreatic islet regeneration and restoration of normal glycemic control in fatally damaged mice using intravenous delivery of an adult progenitor cell type. Surprisingly, xenogeneic PDP cells promoted the regeneration of pancreatic β-cells of host origin. They did not restore normoglycemia through building fully differentiated xenogeneic tissue (i.e., rat or human islets in a diabetic mouse host). Thus, the PDP cells must activate some host cell type, or types, to drive pancreatic islet regeneration, and these cells are not capable of this in the absence of PDP cells. Thus, it is reasonable to propose that PDP cells provide a stimulus for repair. It is remarkable that this effect can be generated in a xenogeneic setting, using human and rat cells, and is sustained for up to 100 days after cell injection. This repair also results in gain and maintenance of body weight in PDP cell-treated diabetic animals, consistent with restoration of pancreatic function. These observations are congruent with recent reports detailing bone marrow–derived mouse c-kit-expressing stem cells initiating endogenous pancreatic tissue regeneration in vivo 16 –18 and with stimulation of endogenous neural repair using exogenous stem cells in mice. 19 However, the mechanism of the induced repair has not been elucidated in either instance. Significantly, in this STZ model of pancreatic damage, tissue regeneration appears to be mediated by developmental recapitulation of insulin gene expression.
The presence of the insulin gene transcripts of both species together with the FISH data indicate that a small number of PDP cells appear to have differentiated to generate insulin-producing cells, but that this is within the context of a massive islet regeneration response of the murine tissue, stimulated by the PDP cells. We made similar observations on efficacy using both CD90 subpopulations of PDP cells, despite only the CD90+ population showing differentiation into an islet-like morphology in vitro. 11 CD90− cells showed no morphological changes, but did express insulin transcripts. Clearly, the in vitro islet differentiation potential of these cells bears no relation to their efficacy for in vivo repair stimulation. It is possible that the repair-active population is a further subpopulation common to both CD90+ and CD90− fractions of the PDP cell population, or that all PDP cells are equivalent in this regard, but this remains to be determined.
The nature of the mouse cell type or types that respond to the presence of PDP cells in the damaged pancreas remains unknown. Potential candidates include functionally analogous murine PDP cells, islet-derived adult stem cells, ES-like cells surviving from embryonic development, activated Ngn3-positive facultative progenitors, other endocrine or exocrine cells of the pancreas, and β-cells that have survived STZ treatment. 20 The latter is unlikely: First, protection of existing β-cells by PDP cells, would not be expected to generate Ins I as well as Ins II transcripts; second, a high dose of STZ was employed and the conditioned medium controls showed no evidence of developing a capacity to control blood glucose. However, it is still formally possible that a combination of mechanisms is occurring simultaneously to produce our observations.
It is important to note that whichever cell is the target of PDP cell action, either directly or indirectly, the cells are incapable of independent regeneration under these experimental conditions, so that the previous data on β-cell regeneration are not necessarily informative.
However, there are several published instances of modes of pancreatic β-cell generation, which must be considered. First, Dor et al. 3 have produced data indicating that the predominant source of β-cell regeneration, following a particular form of injury—50%–70% pancreatectomy—is pre-existing β-cells. Our data are not inconsistent with this observation, but PDP cells must provide significant activation if such a mechanism is operative. Second, Xu et al. 8 reported that Ngn3 facultative endodermal precursors can initiate an embryonic mode of β-cell generation in the adult pancreas after injury by partial duct ligation. Notably, our data, especially the detection of Ins I and Ins II transcripts, are consistent with these observations, but PDP cells must still provide a stimulus. It is possible that the xenogeneic PDP cells initiate this process in host Ngn3-positive cells, or in other cell targets in the pancreas. Third, Zhou et al. 20 have shown that delivery of a combination of three transcription factors (Ngn3, Pdx1, and Mafa) reprograms differentiated pancreatic exocrine cells in adult mice to cells closely resemble β-cells. This is also a potential mode of action of PDP cells, via the activation of appropriate signaling pathways through cell–cell contact and/or soluble factors.
In our experiments, the presence of the Ins I transcript in regenerating pancreas is indicative that endogenous repair processes involve developmental reprogramming in the pancreas. Whether this includes any of the above, or the epithelial–mesenchymal–epithelial transition described for β-cells 21 as part of this process, is undetermined. Fourth, Thorel et al. 22 have demonstrated that in a transgenic model employing chemically destroyed adult pancreas, pancreatic regeneration was via direct reprogramming of pancreatic α-cells. Our data are again, not inconsistent with these data. Fifth, our data are consistent with reports detailing bone marrow–derived c-kit-positive stem cells initiating endogenous pancreatic tissue regeneration in vivo 15,17 and with stimulation of endogenous neural repair using exogenous stem cells in mice. 18 However, the mechanism of the induced repair has not been elucidated in either instance.
Hasegawa et al. 18 have demonstrated that a bone marrow transplant (BMT) can improve hyperglycemia in STZ-induced diabetic mice via regeneration of recipient pancreatic β-cells, consistent with the observations of Hess et al. 16 Interestingly, these authors observed that the population of post-BMT islets was located near pancreatic ducts and blood vessels. However, it appeared that infusion with bone marrow cells, without preirradiation, did not stimulate endogenous regeneration. Whereas irradiation alone had no effect on hyperglycemia, or β-cell regeneration, Hasegawa et al. also observed that BMT had virtually no effect on blood glucose levels or pancreatic insulin content in Nos3−/− transplanted mice, in which mobilization of BM-derived cells is impaired. 18 This raises the hypothesis that immature BM-derived cells can send signals triggering β-cell regeneration. This is consistent with our finding that endogenous pancreatic cells are stimulated to regenerate by PDP cell administration.
Such a mechanism of PDP cell action is important because it further supports the view that the pancreas has a latent, endogenous capacity to regenerate β-cells. Whether this comprises stem-like progenitor cells or reprogramming of other pancreatic cells remains to be demonstrated. These mechanisms are not mutually exclusive, with the cellular substrate for repair possibly being determined by the nature and extent of pancreatic injury. The presence and involvement of a pancreatic stem/progenitor cell is intuitive, based on action of such cells in other organs.
Our data demonstrate clearly that PDP cells, delivered via the bloodstream, can locate to the damaged pancreas and stimulate the remaining tissue to regenerate. The primary effect of PDP cells thus appears to be stimulation of endogenous tissue repair, but a small proportion of this cell population, which may represent a particular subset of cells, differentiates into insulin-producing cells.
The action of PDP cells has further implications for future treatment of autoimmune diabetes. The cells, despite being used in a xenotransplantation setting, appear to be immunologically null. Any ability of such cells to manipulate host immune responses to engender tolerance to their presence is intriguing. This is, however, not without precedent. Paracrine interactions between mesenchymal stem cells (MSCs) and innate and adaptive immune cells (dendritic cells and T lymphocytes, respectively) are well documented. 23 MSCs can modulate T cell–mediated immunological responses, and systemically administered MSCs home to sites of ischemia or injury, analogous to PDPs. This has implications for type I diabetes, where there will be a continual immune attack on any regenerated tissue. It remains to be proven how PDP cells will function in such an environment.
The use of direct in vivo delivery, without the need to differentiate under stressful in vitro conditions, should reduce the risk of malignant transformation, as does endogenous repair stimulation, in contrast to stem cell-derived tissue building. Amounts of insulin sufficient to normalize blood glucose levels are produced and sustained by the repaired pancreas. Consequently, we believe that the use of PDP cells as a vehicle for regenerative medicine offers exciting prospects for future therapies.
Footnotes
Acknowledgments
This research was supported by Darlinda's Charitable Trust through unrestricted awards to P.G.S. and by Scottish Enterprise through an unrestricted award to P.G.S. and R.W.D. The authors would like to thank Drs. Dagmara McGuinness and Christopher McGlynn for assistance with the production of figures.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.