
Abstract
Cancer in elderly patients is an increasingly common problem. Older patients have more co-morbidity, therefore the toxic effects of chemotherapy treatment are less tolerable compared to younger patients. Platinum-based compounds (PBCs) are commonly used cytotoxic agents in the treatment of several solid tumors; however, their application is still limited in elderly patients, due to the risks in cardiovascular toxicity. The increased risk for myocardial ischemia, stroke, and vascular thrombosis linked with PBCs treatment is mainly due to reactive oxygen species (ROS) production and the subsequent induction of oxidative stress and switch to a prothrombotic condition. Recently, studies have shown a different genetic susceptibility in cardiovascular toxicity induced by therapy with PBCs. Antioxidants, such as vitamin E, selenium, lycopene, melatonin, and resveratrol, have been implicated in cancer treatment by their property to suppress the oxidant injury. Resveratrol, especially, has been shown to increase the antineoplastic activity of cisplatin. In addition, resveratrol's ability to activate the sirtuin1 (SIRT1) pathway has been heavily implicated in the mechanisms controlling longevity and quality of life in the aged population. This article reviews the current state of treatment with PBCs and their associated risk for cardiovascular disease. It discusses the most powerful antioxidant supplementation options as a possible strategy to reduce the cardiovascular toxicity effects of chemotherapy in the elderly.
Introduction
Cisplatin (cis-diamminedichloroplatinum II or CDDP) is a critical component of therapeutic regimens in a broad range of malignancies, including non–small cell lung cancer 4 –6 and breast, 7 testicular, 8 and ovarian carcinomas. 9,10 Nonetheless, its administration is associated with significant toxicity, particularly in the elderly, with compromised performance status, decline of renal function, and co-morbidities that may preclude the safe administration of cisplatin to a significant proportion of patients. In attempts to overcome this problem, the development of platinum-based drugs has progressed over the years to carboplatin and oxaliplatin. Compared to cisplatin, carboplatin causes lower rates of nephrotoxicity and neurotoxicity, thus representing an appealing alternative for cisplatin-based chemotherapy, especially in elderly. Nonetheless it has been recently suggested that cisplatin-based chemotherapy in elderly patients should be a priority over carboplatin evaluation 11 on the basis of both the results of a recent meta-analysis 12 and the consideration that, despite reduced renal and neural toxicity, carboplatin has a higher rate of hematological toxicity than cisplatin, particularly when combined with other mielotoxic drugs as gemcitabine. 11
Thus, in elderly patients, safety also remains a problem with carboplatin-based chemotherapy, although advanced age alone should not preclude platinum-based chemotherapy. 11,13,14 However, it must be noted that selection bias might have influenced all of these trials, because elderly patients were likely not representative of the whole elderly population, but rather of a small subgroup thought by investigators to be eligible for aggressive treatment. 10 Moreover, clinical trials specific to the elderly population have mostly been small prospective phase I/II trials or retrospective analyses. 13 –17 Therefore, prospective clinical trials of platinum-based chemotherapy, with inclusion criteria limited to the elderly population, are still needed.
More than 30 years after its clinical benefits were first recognized, the exact mechanism of cisplatin action is still not well understood. Although there is no doubt that DNA is the primary target of cisplatin, there are still gaps in our comprehension of the process that translates cisplatin-induced DNA damage into its characteristic drug-mediated cellular effects—namely, inhibition of DNA synthesis, suppression of RNA transcription, effects on the cell cycle, and the therapeutically beneficial process of apoptosis. 18 Presently, it is generally acknowledged that cisplatin binds to DNA, leading to the formation of inter- and intrastrand cross-links, which results in defective DNA templates and arrest of DNA synthesis and replication. 19,20 The molecular mechanisms linking the formation of DNA adducts to cell death–inducing signaling are not well understood, but at least two apoptotic signaling pathways may be implicated. One involves calpain activation and calpain-mediated cleavage of the proapoptotic BH3-only protein Bid. 21 The other one results in MEKK11-dependent modulation of the proapoptotic protein Bak. 22
Nevertheless, even though DNA adducts are considered the key toxic lesions induced by cisplatin, only 5–10% of covalently bound cell-associated cisplatin is found in the DNA fraction, whereas 75–85% of the drug binds to proteins and other cellular constituents. 23 When cisplatin passes through the cell membrane, it may associate with some constituents of the lipidic bilayer, which contain nitrogen and sulfur atoms, including phospholipids and phosphatidylserine. Thus, cisplatin might induce apoptosis in the absence of DNA damage, and the endoplasmic reticulum may represent its nonnuclear target. 24 In addition, recent studies have reported evidence that cisplatin may cause apoptosis in cancer cells through a disruption of the mitochondrial transmembrane potential that precedes nuclear DNA fragmentation, 25 probably through dysregulation of the uncoupling protein-2 (UCP2) and increased production of mitochondrial reactive oxygen species (ROS), inhibition of mitochondrial respiration, and decreased mitochondrial energy competence to produce adenosine triphosphate (ATP). 26 On the other hand, one of the most important non-DNA targets of cisplatin is probably glutathione (GSH). GSH, together with other thiol-containing biomolecules, such as metallothioneins, binds quickly to platinum and, as a result, this negative pharmacological property affects the cell's development of resistance and toxicity. 23
Collateral Effects of Platinum-Based Compounds: Risk for Cardiovascular Disease in the Elderly
The toxic effects of PBCs in human and animals include nephrotoxicity, ototoxicity, and neurotoxicity, whose general, pathological and molecular aspects have been reviewed extensively. 27 –29 Cardiotoxicity and thromboembolic complications (i.e., myocardial infarction, pulmonary embolism, and stroke), however, have been reported occasionally. 30 In this respect, it is imperative that there be a rigorous evaluation of the risk-versus-benefit ratio in elderly patients, in whom reduction of creatinine clearance and cisplatin renal excretion is expected to increase the potential for toxicity. Furthermore, age-related presence of co-morbidities and compromised performance status may preclude the safe administration of PBCs in elderly patients with preexisting cardiovascular risk. 6
The development of thromboembolic disease is dependent upon the relationship between the factors of Virchow's triad—stasis, hypercoagulability, and vessel wall injury. As first described by Trousseau in the nineteenth century and supported by contemporary studies, some patients with malignancy are in a hypercoagulable state and do develop thromboses. 31 The etiology of cancer-associated thrombosis is likely multifactorial, with different factors assuming various degrees of importance, depending on clinical setting, patient characteristics and co-morbidities, or therapeutic interventions. 31,32 Chemotherapy, in fact, is associated with a two- to six-fold increased risk of thromboembolic events as compared to the general population. 33 This may be the result of an activation of hemostasis, occurring via vessel wall damage or changes in the clotting cascade. 31
Independently of the clinical–pathological features, the risk of suffering an arterial or venous thromboembolic complication during the course of cisplatin-based chemotherapy might be as high as 10–20%. 34 Most studies show a two-fold relative risk of cardiovascular disease (CVD) compared with the general population and also an increased mortality rate from cardiovascular events. Additional risk factors, such as smoking and family history of coronary artery disease, could also increase the likelihood of cardiac events. 34 However, data on the risk of vascular complications after cisplatin chemotherapy are limited, because clinical trials often do not specify the incidence and severity of treatment-related cardiovascular (thrombotic) toxicities. As a corollary, Anders et al. recently highlighted the remarkable difference in incidence of severe or life-threatening cardiovascular toxicity observed among various studies, suggesting that vascular complications were inadequately reported. 35 However, the concomitant use of antiangiogenic and/or supportive drugs (i.e., erythropoietin or corticosteroids) must also be taken into consideration, because they alone may contribute to the increased risk of thromboembolic complications. 35 –37
Among cisplatin-related vascular toxicities, cerebrovascular accidents, such as stroke or thrombosis of the internal carotid artery, have been reported as the most common complication, often affecting young patients. 38 –46 Other clinical manifestations include coronary ischemia, such as acute chest pain syndromes, palpitations, and myocardial infarction. 47 –51 Moreover, a retrospective study by Meinardi and colleagues demonstrated a significant increase in cardiac events, as well as an unfavorable cardiovascular risk profile in long-term survivors of metastatic testicular cancer treated with cisplatin. In addition, a delayed appearance of cardiovascular complications, such as hypertension, left ventricular hypertrophy, myocardial ischemia, and infarction was associated with cisplatin use. 52 Cisplatin CVD complications were recorded as late as 10–20 years after remission 52 and were related to the cumulative chemotherapy dose. 53
Despite more than 30 years of clinical use, no definite mechanism has been proposed for the cardiovascular toxic effects of cisplatin, but clinical and experimental studies have suggested a number of potential mechanisms that might account for vascular complications of platinum-based therapies, which are reviewed next.
Platinum-Based Compounds and Vascular Toxicity: Risk Mechanisms for Thrombosis
Endothelial dysfunction
Among the possible causative mechanisms of cisplatin-related acute vascular complications, dysfunction and/or damage of endothelial cells may play a pivotal role. This is further demonstrated by an elevation of the von Willebrand factor antigen, which has been implicated in the occurrence of hemolytic–uremic syndrome, thrombotic thrombocytopenic purpura, and cerebrovascular accidents in older patients treated with cisplatin. 44 The prevalence of the Raynaud phenomenon, 49 as well as anecdotal evidence of coronary spasm in otherwise normal vessels, 47 further corroborates this hypothesis. Furthermore, chronic toxicity might be caused by renal endothelial damage as well, leading to delayed hypertension and microalbuminuria. 51 Cisplatin may exert deleterious effects not only on the endothelial layer but also on basement membrane and capillary pericytes. 54 This action occurs through increased endothelial lipid peroxidation 43 or activation of pro-apoptotic signaling pathways. 55
Platelet activation
Increased platelet aggregation 56 and/or enhanced thromboxane (Tx) formation by platelets 57 have been also implicated, although in vitro studies on animal and human models have yielded conflicting results. Indeed, some authors failed to demonstrate any direct effect of cisplatin on platelet function tests 58 –60 and suggested that the effects of cisplatin depend on the monocytes to produce platelet aggregation, which is mediated by a P-selectin–dependent mechanism. 61 In vitro studies have shown that cisplatin is able to induce in vitro activation of the arachidonic acid (AA) pathway in human platelets (increasing human platelet reactivity) by activating platelet phospholipase A2 (PLA2). 57 This finding is further corroborated by clinical data showing that cisplatin administration causes increased urinary excretion of Tx metabolites in ovarian cancer patients. 62 Finally, coagulation activation has been also demonstrated during the course of cisplatin-based regimens, possibly related to increased tissue factor expression by monocytes 63 or microparticle release during the second or third treatment cycle, 64,65 when vascular toxicity is mostly evident. 39,66
Oxidative damage
Despite the apparent differences, all of these mechanisms appear to share a common pathogenetic root—the cisplatin-induced generation of ROS. ROS is a collective term referring to oxidizing agents, including free radicals and certain nonradicals, which might be turned into radicals. Their production is normally controlled both in health and disease by the antioxidant defense mechanism that includes intracellular enzymes (i.e., glutathione peroxidase and superoxide dismutase) and low-molecular-mass compounds (i.e., vitamin E). Although repair mechanisms are effective, some steady-state basal oxidative damage still occurs in all individuals. Oxidative stress arises when there is a marked imbalance between the production and removal of ROS. This may originate from an overproduction of these substances or from depletion in the antioxidant defenses, such as in aging.
Certain drugs may induce oxidative stress by forming drug-derived radicals that cannot only deplete the antioxidant defenses, but can also react directly with biomolecules. In this respect, there is some evidence to suggest that free radical–mediated damage may be involved in the toxicity of PBCs. 67 Although there is no known pathway by which these drugs generate ROS directly, it has been proposed that oxidative stress is an important mechanism of cisplatin-induced normal tissue toxicity (Fig. 1), 68 –74 but not of carboplatin, 75 which might explain the lower rate of toxicity with respect to its first-generation analog and justify the choice of carboplatin over cisplatin in the elderly. Nonetheless, the possibility that cisplatin and carboplatin may share the same ROS-based mechanisms of toxicity has been recently raised, based on the findings that carboplatin can induce cardiotoxicity in animal models and in cultured cells via a mitochondrial pathway related to ROS production, 76 a mechanism that might be counteracted by pravastatin administration. 76,77
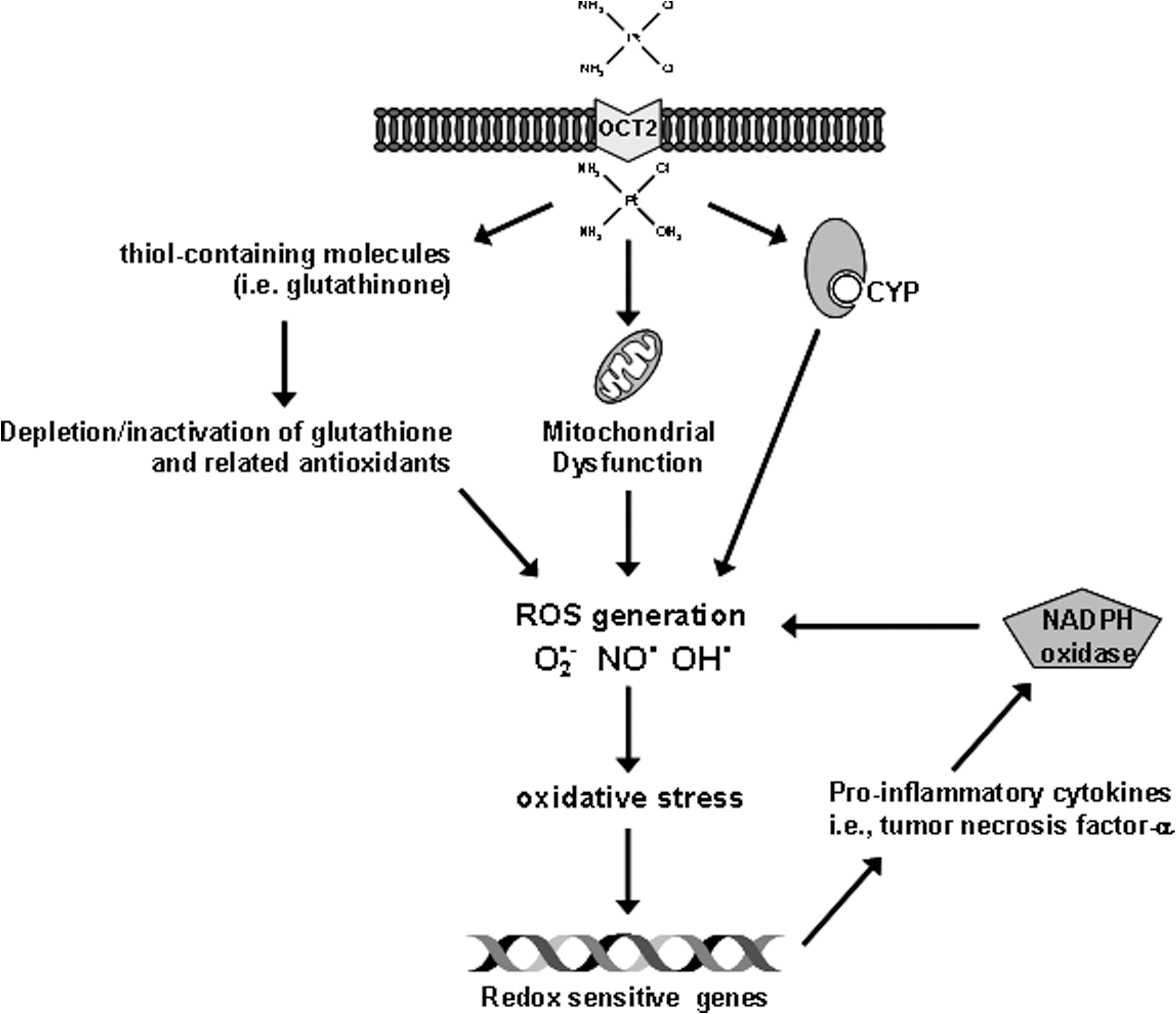
Proposed mechanisms of reactive oxygen species (ROS) cisplatin-induced generation. 1 Cisplatin enters into cells by organic cation transporters (OCT2) and is aquated into a highly reactive form, which can react rapidly with thiol-containing molecules including glutathinone, a well-recognized cellular antioxidant. Depletion or inactivation of glutathione and related antioxidants by cisplatin is expected to shift the cellular redox status, leading to the accumulation of endogenous ROS and oxidative stress within the cells. Second, cisplatin may induce mitochondrial dysfunction and increase ROS production via the disrupted respiratory chain. Third, cisplatin may induce ROS formation in the microsomes via the cytochrome P450 (CYP) system, which is an important source of catalytic iron for ROS generation during cisplatin treatment. Secretion of proinflammatory cytokines through activation of redox-sensitive genes might be responsible for an increase of icotinamide adenine dinucleotide phosphate (NADPH) oxidase activity through NOX1 and NOX4 mRNA expression and NOX subunit p47phox and p67phox proteins, with additional ROS production. 46 O2 •−,superoxide anion; NO•, nitric oxide radical; OH•, hydroxyl radical.
In addition to an increased ROS production, it has been shown that cisplatin-based combination chemotherapy induces a fall in plasma antioxidant levels that may reflect a failure of the antioxidant defense mechanism against oxidative damage induced by cisplatin. This probably results from consumption of antioxidants caused by chemotherapy-induced oxidative stress as well as renal loss of water-soluble, small-molecular-weight antioxidants. 78 More recently, it has been suggested that the modulation of sirtuin 1 (SIRT1) expression constitutes a molecular tool for the prevention of cisplatin-induced toxicity. 79 SIRT1, a nicotinamide adenine dinucleotide+ (NAD+)-dependent protein deacetylase, might exert protective effects on the vascular endothelium by regulating the endothelial nitric oxide synthase (eNOS) and by preventing stress-induced endothelial dysfunction. 80 In addition to its cell-autonomous role in the vascular endothelial homeostasis, SIRT1 is also involved in the preservation of peroxisome numbers and function impaired in cisplatin-related toxicity, concomitant up-regulation of catalase, and elimination of excessive ROS. 79
Cisplatin-induced oxidant stress may also cause lipid peroxidation, 81 although this does not appear to represent a major contributor to cisplatin-induced cell death. 73 Nonetheless, lipid peroxidation might contribute to platelet activation by generating bioactive F2-isoprostanes, which may act as the primary trigger of thromboxane-dependent platelet activation 82 and may promote endothelial dysfunction through increased production of ROS, inactivation of nitric oxide (NO), and activation of the nuclear transcription factor NF-κB. 83,84
The relationship between ROS and vascular function has been extensively investigated, because an increased production of ROS and NO impairment are key features of vascular dysfunction. 85 In addition to the effects mediated by NO scavenging by superoxide anion, increasing evidence suggests that the peroxynitrite formed by the superoxide–NO interaction, has detrimental effects on vascular function due to oxidation of cellular proteins and lipids 85 or direct cell toxicity. 86 Moreover, superoxide–NO interactions may play a role in the modulation of vascular signaling through redox-sensitive mechanisms capable of activating key molecules, such as protein kinase C (PKC) and NF-κB, which are responsible for transcription of genes encoding cytokines, growth factors, adhesion molecules, and a consequent switch toward a condition of endothelial dysfunction and a prothrombotic condition. 87
In this respect, a particular emphasis has been given to the interactions occurring between vascular cells and platelets. Platelets represent a prime target for ROS produced or released in the vascular lumen, and the results obtained suggest that low levels of oxidants may promote aggregation, whereas exposure to higher, albeit no-toxic concentrations of exogenous hydrogen peroxide (H2O2), may result in platelet inhibition. 82 As stated above, increased in vitro and in vivo platelet activation has been related to cisplatin administration, 57,59,62 which was also directly shown to induce changes in the biological activity of blood platelets, including increased platelet lipid peroxidation and superoxide generation, 88 probably through a glutathione-associated metabolism of PBCs. 89
On the contrary, the clinical situation is clearly more complex, because the net effect of ROS on intravascular thrombosis is also dependent on the integrity of the endothelium as well as on oxidant-mediated alterations of other major players of thrombosis, such as coagulation factors. In addition to their direct effects on platelet function, ROS may cause enhanced activation of the coagulation cascade, ultimately leading to thrombin formation, via their combined effects on stimulation of tissue factor (TF) activity and inhibition of fibrinolytic pathways. 82,90 Indeed, a brief period of exposure to ROS resulted in a significant increase in TF mRNA levels, accompanied by the appearance of large TF procoagulant activity, 91 an effect that was abolished by oxygen radical scavengers 91 or NO. 92 Besides this effect, ROS may also promote intravascular thrombus formation by interfering with mechanisms that normally inhibit activation of the coagulation pathway. For example, lipid peroxides, formed as a consequence of ROS attack to circulating lipoproteins, can increase the amount of thrombin produced and can slow down the rate of thrombin decay due to inhibition of plasma antithrombin. 93 Similar susceptibility to ROS-mediated inactivation has been reported for other key antithrombotic factors, such as alpha-2-antiplasmin, 94 plasminogen activator, 95 and thrombomodulin. 96 Moreover, it has been shown that the reaction of peroxynitrite with fibrinogen results in both structural modifications and altered biological properties of this glycoprotein, thus impairing its ability to mediate platelet adhesion and aggregation. 97
Antioxidants: Antiaging Properties and Potential Co-Adjuvant Therapy
Aging has been defined as the accumulation of diverse deleterious changes in the cells and tissues that increase the risks of disease and death. 98 This theory is a continuation of Harman's first theory of aging, where in the 1956 he argued that: “the aging process is determined by the sum of the deleterious free radical reactions occurring continuously throughout the cells and tissues of organisms.” 99 Currently, the free radical theory is most described and accredited. It has been further supported by new discoveries of ROS production and implication in causative cellular damage and disease onset. 100
ROS are able to damage cellular components directly or lead to the generation of secondary reactions, which then initiate oxidative processes. ROS include a number of chemically reactive molecules derived from oxygen, such as H2O2, superoxide anion, and the hydroxyl radical, that are able to interact destructively with DNA, proteins and lipids. 101
For many years, in vitro and in vivo studies aimed to investigate the antiaging properties of natural compounds that might reduce this oxidative damage. Several antioxidants have been shown to have beneficial effects on aging by activating different pathways, leading to a reduction in ROS production or protection against ROS damage, and therefore protection against typical diseases of aging, such as CVD. Oxidative stress promotes the development of coronary artery disease and stroke through several mechanisms. These mechanisms include: (1) Endothelial dysfunction, (2) vascular inflammation, and (3) increase in arterial stiffness, where in addition to oxidative stress mediated mechanisms, hormonal and genetics factors have been shown to play a pivotal role. 102
Figure 2 shows the vascular damage and toxicity caused by PBCs and the most potent antiaging/antioxidants available to reduce vascular toxicity. Antioxidants have been implicated in cancer prevention and therapy for their capacity to protect against free radicals damage. 103 Many compounds have been studied in both fields, CVD in aging, and cancer, such as vitamin E, lycopene, selenium, melatonin, and resveratrol, among others. 103,104 Table 1 shows that natural compounds, through their antioxidants properties, have been demonstrated to counteract PBCs toxicity. 103 Some of the most important clinical trials aimed to test the effect of the antioxidants supplementation as coadjuvant to reduce PBC toxicity are listed in Table 2. The results of human clinical trials in demonstrating the protective effect of antioxidants against toxicity caused by chemotherapy were often controversial. Some trials have shown beneficial effects of antioxidant supplementation for compounds such as melatonin, whereas other trials have failed to show a reduction in PBCs-induced toxicity (Table 2). There are many potential explanations for these disparities—different types of cancers, different study populations, treatments, and study design, among others.
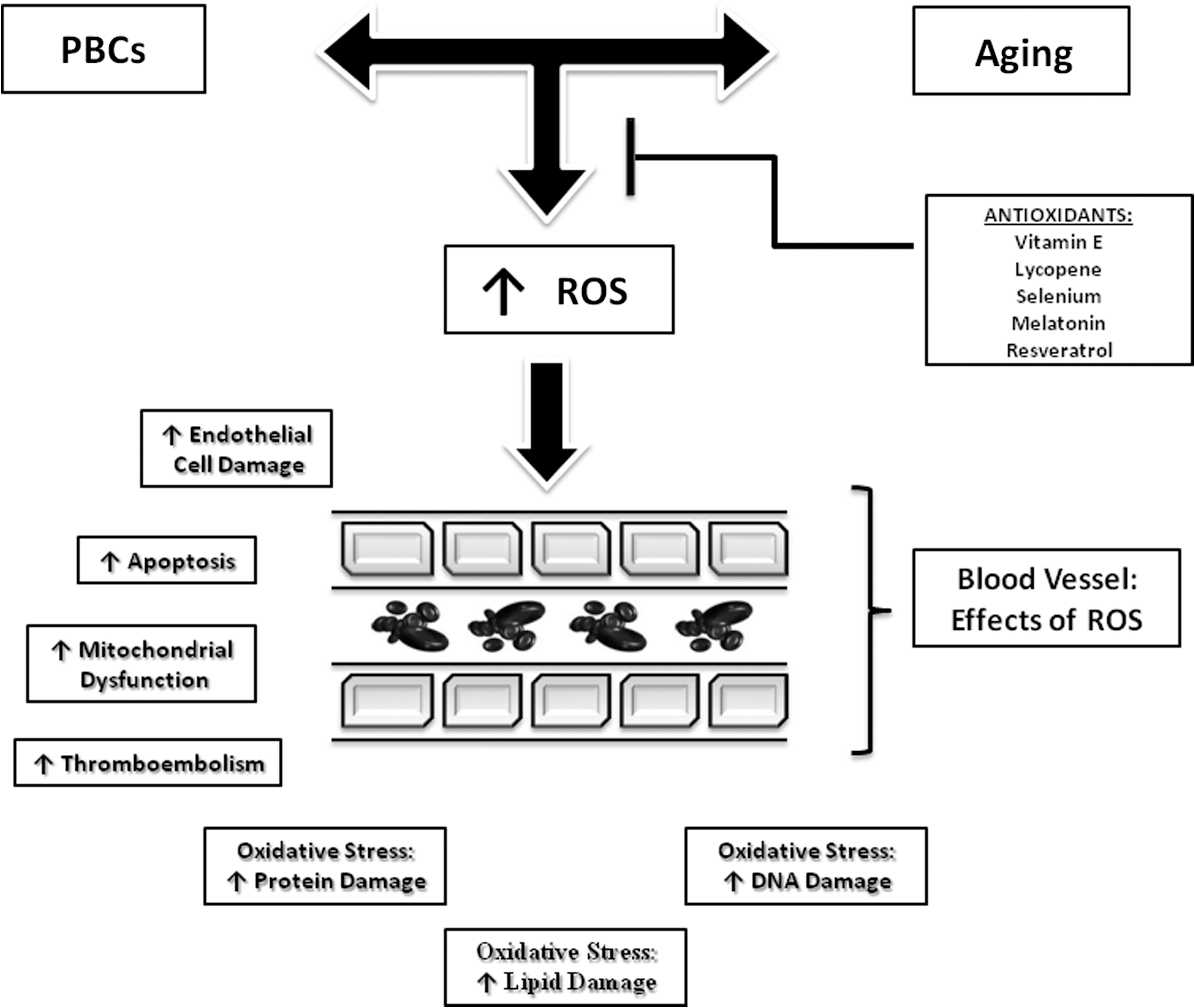
Aging and platinum-based compounds induce vessel damage and toxicity through reactive oxygen species (ROS) production. ROS production leads to several mechanisms inducing vessel damage. Antioxidants may block these injury mechanisms by reducing ROS production. PBCs, Platinum-based compounds.
5-LOX, 5-lipoxygenase; AKT, serine/threonine protein kinase; AP-1, activator protein 1; BAX, Bcl-2–associated X protein; Bcl-2, B-cell lymphoma 2; cAMP, cyclic adenosine 30, 50-monophosphate; CD8+, cluster of differentiation 8; CDK, cyclin-dependent kinase; COX-2, cyclo-oxygenase-2; cyclin D1, G1/S-specific cyclin-D1; DAG, diacyl glycerol; DGK, diacylglycerol kinase; DR, death receptor; EGFR, epidermal growth factor receptor; eNOS, endothelial nitric oxide synthase; ERK, extracelluar signal–regulated kinase; Fas, Fas death domain (TNF receptor superfamily, member 6); HMG-CoA reductase, 3-hydroxy-3-methyl-glutaryl-coenzyme A; IGF-1R, insulin-like growth factor-1 receptor; IGF-BP3, insulin-like growth factor binding protein 3; IL-1β, interleukin-1β; IL-12, interleukin 12; IL-6, interleukin-6; iNOS, inducible nitric oxide synthase; JNK, Jun-N-terminal kinase; MAPK, mitogen-activated protein kinase; MEK, MAP ERK kinase; MMP-2/9, matrix metalloproteinases; NF-κB, nuclear factor-κB; NO, nitric oxide; PAI-1, platelet activation inhibitor-1; PDGF, platelet-derived growth factor; PKA, protein kinase A; PKB, protein kinase B; PKC, protein kinase C; PKG, protein kinase G; PLA2, phospholipase A2; PLC, phospholipase C; PP2A, protein phosphatase 2A; PPAR-γ, peroxisome proliferator-activated receptor-γ; PUMA, p53 up-regulated modulator of apoptosis; RAS, Rat Sarcoma; Sirt 1, sirtuin (silent mating type information regulation 2 homolog) 1; SOD, superoxide dismutase; Sp, stimulating protein; STAT, signal transducers and activators of transcription; TNF-α, tumor necrosis factor-α; VEGF, vascular endothelial growth factor.
i.v., Intrravenous; p.o., per os; IU, International Units; b.i.d., twice a day.
Vitamin E
Several experimental studies showed that supplementation with vitamin E was associated with reduction of atherosclerosis. 105 The main protective mechanism was attributed to its capacity to protect high-density lipoprotein (HDL) and low-density lipoprotein (LDL) from peroxidation by free radicals. 106 Other mechanisms mediated by vitamin E, such as inhibition of monocyte release of ROS and interleukin-1β (IL-1β), interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α) (all inflammatory cytokines), as well as inhibition of platelet adhesion and aggregation have been demonstrated to play a role in reducing atherosclerosis and aging processes. 106 Recently, a study conducted in C57BL/6 mice reported that lifelong dietary vitamin E supplementation significantly increased median life span by 15%. 107 The author suggested that this increase in life span may reflect an anticancer effect via induction of the P21 signaling pathway by vitamin E. Despite these findings, there is still controversy regarding the benefit of vitamin E supplementation for CVD prevention in clinical trials. 106 Vitamin E has been associated with prevention of neuropathy, 108 nephrotoxicity, 109 and ototoxicity 110 induced by cisplatin. All beneficial actions are mediated by reducing the cellular damage caused by ROS. Vitamin E has also been demonstrated to enhance the anticancer effect of cisplatin. 111
Lycopene
Lycopene is a bioactive red-colored pigment naturally occurring in plants. 112 For its antioxidant properties, both epidemiological studies and human supplementation trials have shown a reduction in risk for CVD. 113 Among carotenoids, lycopene has been demonstrated to be the most potent antioxidant ranked highest to lowest respectively: lycopene>α-tocopherol > α-carotene > β-cryptoxanthin >zeaxanthin = β-carotene > lutein. 114 Like vitamin E, the most important mechanism mediated by lycopene against atherosclerosis and CVD is to prevent LDL oxidation. 115 Lycopene, together with the other natural antioxidants, has been proposed as antiaging treatment. 116 Particularly, lycopene has been shown to attenuate both IL-1β–stimulated and spontaneous human aortic endothelial cell (HAEC) adhesion to U937 monocytic cells, indicating a protective role in endothelial dysfunction. 117 Lycopene has also been demonstrated to prevent cisplatin-induced testicular apoptosis, histopathological lesions, and lipid peroxidation in male rats. 118 Moreover, new findings reported that lycopene mitigates the nephrotoxic effect of cisplatin in rats through Nrf2-mediated induction of heme oxygenase-1 (HO-1). 119 Interestingly, cisplatin-treated animals submitted to acute and subacute treatments with different lycopene doses, showed a significant reduction in the number of abnormal chromosome metaphases, when compared with animals treated with cisplatin only. Consequently, the protective effects of lycopene on chromosomal damage induced by cisplatin have been attributed to its antioxidant activity.
Selenium
Selenium is a component of selenoproteins that is obtained from dietary sources, including, cereals, grains, and vegetables. The most important antioxidant function of selenoproteins is the activity of the glutathione peroxidase enzymes, which catalyze the detoxification of H2O2 and organic hydroperoxides. 120 Selenium, is another antioxidant with a potential role in CVD protection through several mechanisms, including increased LDL resistance to oxidative modification, inhibition of prostaglandin synthesis and platelet aggregation, and protection against toxic heavy metals. 121 However, epidemiological studies and clinical trials report controversial findings. Salonen et al. found an increase in cardiovascular morbidity and mortality for individuals with serum selenium concentrations below 45 μg/L compared with individuals with higher concentrations. 122 On the contrary, a clinical trial of selenium supplementation in primary prevention of CVD failed to find an association for selenium supplementation of 200 μg daily with CVD end points during 7.6 years of follow-up. 123
Selenium supplementation has been proposed to play an important role in reducing the mechanisms of aging. Selenium has been shown to block apoptosis induced by ROS by blocking the signal of the apoptosis signal-regulating kinase 1 (ASK1) and stimulating a survival signal for the serine/threonine protein kinase Akt/PKB activity. 124 Selenium has been implicated in cancer therapy. Recently, a conjugate of selenite with diammineplatinum [(NH(3))(2)Pt(SeO(3))(2)] has been proposed as a novel potential anticancer drug for its ability to inhibit telomerase activity. 125 An experimental study demonstrated that antioxidant defense systems, such as glutathione-S-transferase and catalase, that are depleted by cisplatin therapy, were restored to normal by the selenium compound. 126 Therefore, selenium has been involved in the nephrotoxicity, 126 hepatotoxicity, 127 and hematological alterations 128 induced by cisplatin.
Melatonin
Melatonin (N-acetyl-5-methoxytryptamine) is a secretory product of the human pineal gland. The relationship between melatonin, cardiovascular risk factors, and CVD has long been investigated. Melatonin has been shown to protect against hypertension, ischemic/reperfusion injury, atherosclerosis, and cardiac hypertrophy by modulating several pathways all related to reverse oxidative stress–mediated injury. 129 Additionally, melatonin was proposed as antiaging therapy for these same mechanisms. By stimulating the production of natural killer cells and CD4+ cells, which then inhibit CD8+ cells, melatonin has been shown to have the potential therapeutic value to enhance immune function in aged individuals. 130 This property may have a great impact in preventing the toxicity induced by chemotherapy. To date it is known that by activating specific receptors, melatonin can lead to modulation of transcription factors and consequent altered gene expression implicated in the aging processes, such as neurodegenerative conditions. 131 Clinical trials have described its powerful antioxidant properties against cancer. 132 An interesting in vitro study found that melatonin inhibits cisplatin-induced cytotoxicity by directly scavenging ROS and markedly reduces renal cytotoxicity and DNA fragmentation caused by cisplatin. 133 Moreover, melatonin has been shown to ameliorate ototoxicity 134 and immunosoppression 135 induced by cisplatinum.
Resveratrol
Resveratrol (trans-3, 4′, 5-trihydroxystilbene) is of particular importance because it plays a fundamental role in CVD, aging, and cancer. It has been one of the most studied antioxidants since the discovery of the French Paradox. The French Paradox refers to the fact that, in spite of high saturated fat food intake, the French experience fewer CVD events. This paradox is attributed to the French custom of daily drinking of moderate amounts of red wine containing natural antioxidants, such as resveratrol. 136 Resveratrol is a polyphenol found in grapes and wine and described as a cardioprotective agent. 137 Several in vitro and in vivo studies have identified cardiovascular-protective pathways regulated by resveratrol. Its antiischemic properties are due to its capacity to promote vasorelaxation, reduce lipid peroxidation, reduce serum cholesterol and triglyceride levels, decrease platelet aggregation, and therefore decrease atherosclerosis. 138 Recently, in an in vivo study, we demonstrated that treatment with low doses of resveratrol mimicked ischemic preconditioning (IPC) in the rat brain against global cerebral ischemia. This neuroprotection is dependent on the activation of SIRT1. We also demonstrated that resveratrol afforded neuroprotection by decreasing UCP2 levels and increasing mitochondrial ATP-synthesizing efficiency. 139 IPC is an endogenous mechanism of protection conferred by brief periods of ischemia resulting in reduction of the deleterious effects of a subsequent longer-duration no-flow ischemic episode in heart, brain, and other organs. 140 We demonstrated that this protective mechanism is reduced with aging in the heart 141 and the brain. 142 Through its dynamic properties, resveratrol may play a role in prevention of CVD, especially in elderly. Moreover, it has been recently demonstrated that resveratrol increases the life span of experimental animals by activating SIRT1 in a mechanism similar to caloric restriction. 143 The SIRT1-mechanism by which resveratrol may interact with aging, CVD, and PBCs toxicity is shown in Fig. 3
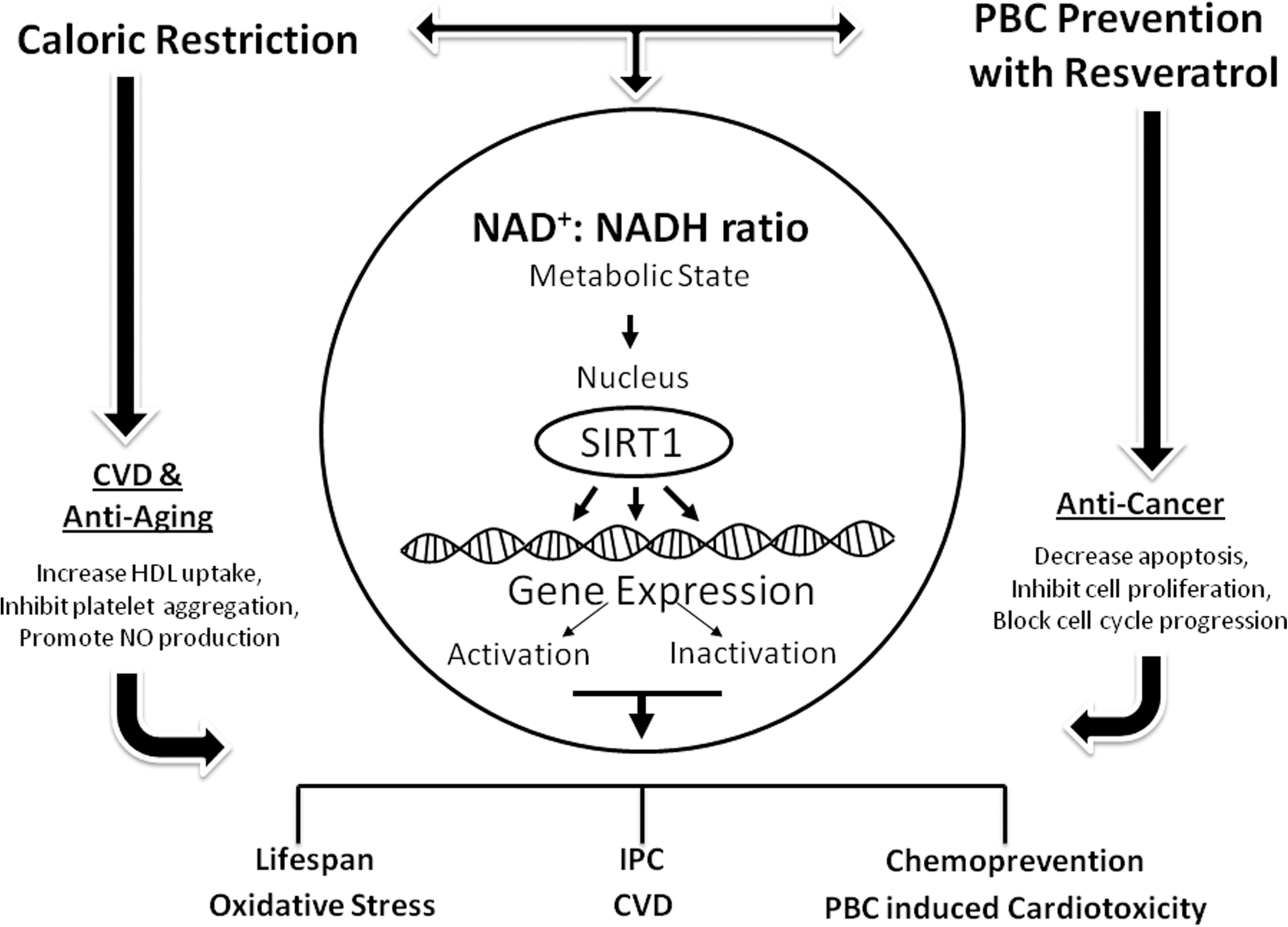
Resveratrol by activating sirtuin1 (SIRT1) has a beneficial effect against aging and cardiovascular disease (CVD) induced by platinum-based compounds. Resveratrol mimics caloric restriction in activating SIRT1 by controlling the metabolic state. SIRT1 activity regulates several transcriptional factors and genetic expression that protect against CVD, aging mechanisms, and cancer. IPC, Ischemic preconditioning; PBC, platinum-based compounds; HDL, high-density lipoprotein.
Recently, the anticancer properties of resveratrol have been identified, and resveratrol has been proposed as a chemopreventive agent. 144 Wang et al. demonstrated that treatment with resveratrol effectively hindered the adverse effects of cisplatin in a dose-dependent manner, such as myocardial injury and impaired heart function via the suppression of oxidative stress. 145 Another study showed that resveratrol prevents apoptosis in normal peripheral blood human lymphocytes during treatment with cisplatinum by protecting from DNA damage. 146 Olas et al. found that resveratrol at different concentrations had a protective effect against oxidative stress in platelets caused by PBCs, and it diminished platelet lipid peroxidation and ROS generation induced by PBCs. Although the promising evidence for the possible use of resveratrol as co-adjuvant in PBCs therapy, the clinical evidence for its use in cancer patients is controversial. Recent findings suggested that alterations in mitochondrial metabolism, mediated by SIRT1, are one of several alterations that can contribute to cellular resistance to cisplatin. 147
Conclusions
PBCs are among the most potent compounds currently used in cancer chemotherapy. However, age-related presence of co-morbidities and compromised performance status may preclude their safe administration in elderly of patients, especially in consideration of the PBCs-related cardiovascular toxicity. The latter has been linked to several cellular pathways, mainly associated with an increase in oxidative stress. Recently, preclinical and clinical studies have led to hypothesizing the protective role of antioxidant in the prevention of age-related disease, as well as PBCs-mediated toxicity. This article has provided evidence regarding the feasibility of combination strategies that might offer protective effects against the toxic risk of PBCs-based cancer therapies, especially in elderly population. Identification of novel approaches that protect physiological homeostasis, without limiting therapeutic effects in cancer, would lead to improvement of PBCs potency and avoidance of its undesired side effects. Modulation of oxidant PBCs-induced stress by the use of antioxidant compounds that interfere with ROS-related vascular damage would improve therapeutic response, reduce toxicity, and constitute a complementary strategy to manage anticancer therapy in the elderly. Further multicenter prospective studies involving large number of elderly patients are warranted to fully establish the real impact of antioxidant compounds in preventing PBCs-mediated cardiovascular toxicity.