
Abstract
Mammalian target of rapamycin (mTOR), a serine/threonine kinase and component of the mTORC1 signaling complex, acts as an energy, nutrient, growth factor, stress, and redox sensor to increase protein synthesis and decrease macroautophagy. mTORC1 plays a central role in the maintenance of homeostasis and its deterioration, seen in aging. The Food and Drug Administration (FDA)-approved immunosuppressive macrolide rapamycin binds immunophilin FKBP12 (FK506-binding protein) to inhibit mTORC1. Unlike most other interventions tested to date, inhibition of mTORC1 by rapamycin extends life span in old mice, likely by a combination of increased autophagy and decreased mRNA translation. Hutchinson–Gilford progeria syndrome (HGPS) is a lethal genetic disorder affecting children that is characterized by symptoms of premature aging, such as atherosclerosis. Increased autophagy induced by rapamycin reduces accumulation of progerin, an alternate spliced form of lamin A/C, that forms insoluble toxic aggregates, resulting in reduced HGPS-associated nuclear blebbing, growth inhibition, epigenetic dysregulation, and genomic instability. Rapamycin-induced autophagy also suppresses symptoms in mouse models of Alzheimer, Parkinson, and Huntington diseases, where toxic insoluble protein aggregates accumulate. On the basis of these results, modulation of mTORC1 function is a promising target for the development of therapeutics for neurodegenerative diseases and HGPS. Rapamycin is the obvious candidate for near-term evaluation in the treatment of these diseases. However, the substantial set of rapamycin-associated adverse effects, as well as the lack of aging-specific human data, should caution the routine use of rapamycin as an antiaging agent. The use of safer, but perhaps weaker, indirect mTORC1 inhibitors, such as metformin and resveratrol, may prove useful. Further study will ascertain whether such compounds extend human health or life span.
Inhibition of Mammalian Target of Rapamycin Extends Life and Health Span
The mammalian TOR (mTOR), a serine/threonine kinase, is found in two signaling complexes, mTORC1 and mTORC2 (see Fig. 1). mTORC1 acts as an energy, nutrient, growth factor, mitogen, stress, and redox sensor and controls protein synthesis and autophagy in a reciprocal fashion. 3 For example, mTORC1 is activated by increased energy and nutrients to positively regulate S6 kinase (S6K) and eIF4e-BP(4E-BP1) to initiate and augment translation. Autophagasomal regulators Unc51-like kinase 1 (ULK1) and ATG13 are inhibited by mTORC1 phosphorylation. Although less well characterized, mTORC2 responds to growth factors to regulate cell polarization via the actin cytoskeleton and cell survival and metabolism via phosphorylation of prosurvival Akt, protein kinase C, and glucocorticoid-regulated kinase (SGK). 4 Akt promotes survival by phosphorylating proapoptotic transcription factors FOXO1 and FOXO3 to inhibit their translocation to the nucleus. Activation of mTORC2 can also contribute to increased activity of mTORC1 via Akt activation of TSC2 and Rheb4 (see Fig. 1). The immunosuppressive macrolide rapamycin, which binds the immunophilin FK506-binding protein (FKBP12), specifically inhibits mTORC1, but not mTORC2 by interfering with the interaction of mTOR with the mTORC1-specific Raptor protein. 3,4 Unfortunately, this picture is muddied in that chronic high doses of rapamycin can also inhibit mTORC2 in some cell types. 6
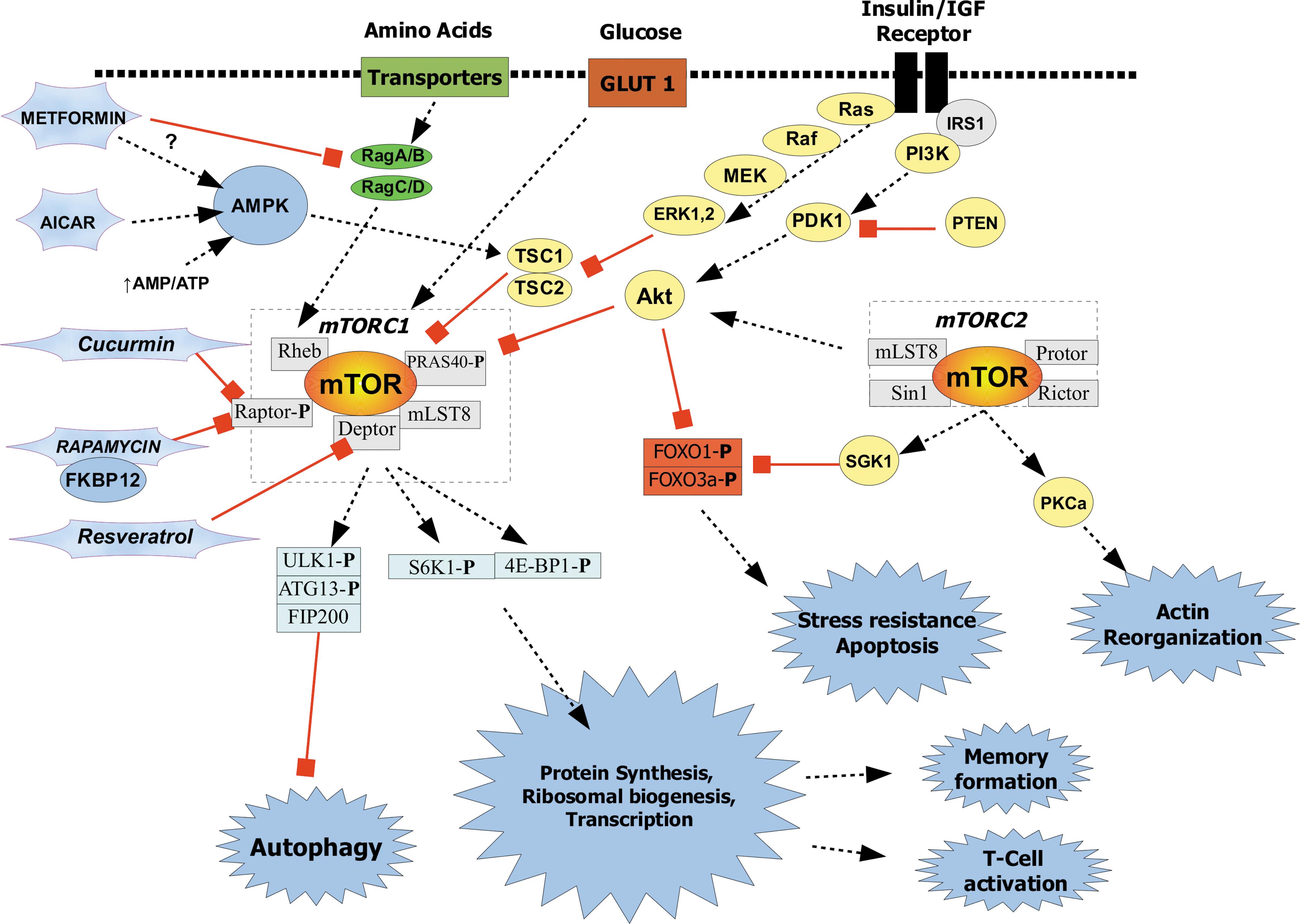
Simplified schematic of mammalian target of rapamycin (mTOR) signaling pathways. mTORC1 promotes messenger RNA (mRNA) translation and inhibits autophagy by integrating growth factor signals like insulin/inslin-like growth factor (IGF), amino acid nutrient signals, and adenosine triphosphate (ATP) levels via 5′-adenosine monophosphate-activated protein kinase (AMPK). mTORC1 also senses DNA damage and hypoxia (not shown). mTORC1 affects many homeostatic processes beyond cell growth and autophagy, including neural memory formation/consolidation and immunity. Growth factors activate mTORC2 to activate Akt and Sgk1, which promote survival and inhibit forkhead transcription factors FOXO1 and FOXO3. The forkhead transcription factors promote antioxidant enzyme expression and cell death. The molecular targets of mTORC1 inhibitors, rapamycin, curcumin, resveratrol, metformin, and 5-aminoimidazole-4-carboxamide-1-β-
Unlike most other interventions, inhibition of mTORC1 by rapamycin extends life span in mice, even when given to old individuals (600 days old). 7 The mechanisms that underlie this intriguing result are under investigation. However, there are two non–mutually exclusive mechanisms that are thought to play an important role, increased autophagy 8 and decreased mRNA translation. Overexpression of Atg8 is known to increase autophagy and extend life span in fruit flies. 2 However, in other model organisms (worms and mice), increased autophagy does not extend life span but appears to be necessary for life extension by other means, such as calorie restriction (CR) and rapamycin. Optimal levels of autophagy are undoubtedly important because high levels can contribute to cell death. In contrast to the autophagy results, reduced translation via knockout of S6 protein kinase 1 (S6PK1) in mice contributes to increased life span and increased resistance to age-related pathologies, such as loss of insulin sensitivity and motor, bone, and immune dysfunction. 9 It is not unreasonable to hypothesize that both mechanisms play an important role in extension of longevity in mammals. 2
Rapamycin Suppresses Hutchinson–Gilford Progeria Syndrome in Tissue Culture
In a fascinating study, Cao et al. 10 show that rapamycin can suppress the phenotype of Hutchinson–Gilford progeria syndrome (HGPS) cells in culture. HGPS is a lethal genetic disorder affecting children who exhibit many characteristics of premature aging, including alopecia, osteoporosis, sclerodermatous skin, and atherosclerosis. Patients with HGPS have an average lifespan of 12 years and typically die from myocardial infarction or stroke. HGPS is caused by a genetic mutation that partially activates a cryptic splice site in exon 11 of the lamin A/C gene, which encodes a nuclear envelope protein. The resulting abnormal protein, progerin, is thought to cause HGPS. 11 Interestingly, progerin is expressed to a small extent in wild-type humans and slowly accumulates with age. Primary fibroblasts from HGPS patients exhibit a variety of abnormal phenotypes, including nuclear blebbing, punctate cytoplasmic accumulation of progerin, 12 reduced growth rate, and decreased Hayflick limit. As might be expected from a protein intimately associated with nuclear function, significant epigenetic changes are associated with HGPS, particularly alterations in histone methylation. Aberrant epigenetic regulation (developmental drift) 13 and accumulation of aggregated proteins 14 are under active investigation as key processes by which cells age. Such work reinforces the relevance of HGPS as a model of premature aging as well as an important syndrome in need of new therapies. 15
Treatment of HGPS cells for 10 days with rapamcyin reduced nuclear blebbing, as determined morphologically by anti-tubulin staining. In addition, the reduced cellular growth rate associated with HGPS was reversed by rapamycin. This was somewhat surprising to the authors because rapamycin itself is known to inhibit cell proliferation. However, the ability of rapamycin to inhibit the rate of cellular growth varies as some cells respond differentially to lower S6K and eIF4e-BP activity. 16 Rapamcyin also delayed replicative senescence by 30 days (≈10 passages) in both normal and HGPS cells, as assessed by senescence-associated β-galactosidase expression and cell number. 10
Accumulating evidence suggests that epigenetic dysregulation plays a major role in aging. 7 Progerin has been shown to progressively alter trimethylation of histone H3 Lys27 (H3K27me3), which marks tightly packed, transcriptionally inactive, facultative heterochromatin. 17 In normal female cells, H3K27me3 coats the inactivated X chromosome (Xi). However, in female HGPS cells, coating of Xi by H3K27me3 is partially lost and is restored by exposure to rapamycin. 10
Genomic instability has also been hypothesized to play a role in aging. HGPS cells exhibit genomic instability, including increased sensitivity to DNA cross-linker mitomycin C and increased expression of DNA-damage associated p53-binding protein 1 (TP53BP1). 18 Rapamycin treatment of HGPS cells significantly reduced anti-TP53BP1 nuclear staining as well as cellular sensitivity to mitomycin C. 10
Because HGPS results from accumulation of progerin, which appears to correlate with nuclear blebbing, 12 the authors hypothesized that rapamycin would lower levels of progerin by increasing its degradation via autophagy. Indeed, rapamycin was seen to reduce nuclear progerin in HGPS nuclei, as well as the half-life of progerin measured by a pulse-chase experiment. Two experiments supported the beneficial role of rapamycin to reduce progerin levels via increased autophagy: (1) Bafilomycin A, a lysosomal pump inhibitor reversed the effect in HPGS cells; and (2) small interfering RNA (siRNA) to ATG7 (known to be essential for autophagy) reversed the effect in a HeLa cell-based progerin expression model. Progerin was found to interact with autophagy adapter protein p62 through K63 ubiquitination and autophagy-linked zinc finger protein FYVE, which functionally links progerin with autophagasomes. 10
Age-associated increases in protein aggregation is another mechanism that may play a major role in aging. 19 Progerin has been shown to aggregate with lamin A to form insoluble complexes that may promote cellular dysfunction. Rapamycin treatment increased the solubility of progerin, by promoting formation of a soluble lamin A progerin A dimer. 10 This may result from the reduced progerin levels found in rapamycin-treated cells. In summary, Cao et al. suggest a central role of augmented autophagy in the beneficial effects of rapamycin, although the potential role of decreased initiation and elongation of translation was not well explored. Cao et al. use these basic science findings to make a strong case in favor of the initiation of clinical trials for treatment of inevitably fatal HGPS.
Rapamycin Suppresses Neurodegenerative Diseases in Mouse Models
Given the beneficial effects of rapamycin on HGPS cells, it would be expected that rapamycin might also benefit other diseases in which clearance of toxic protein aggregates is helpful. Indeed, rapamycin has been shown to inhibit pathology in mouse models of neurodegenerative diseases in which protein aggregation plays a pathological role: Alzheimer, Parkinson, and Huntington diseases. 20 –22 The two mouse models of Alzheimer disease are triple transgenic mice with mutations in PS1, amyloid precursor protein (APP), and tau, and the PDAPP mouse, which carries a mutant APP, rapamycin-extended life span, and reduced β-amyloid levels, plaques, and neurofibrillary tangles. Importantly, the levels of rapamycin used did not impair memory 20 (see below).
In a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinson's disease mouse model, rapamycin helped maintain dopaminergic neuron cell numbers and reduced loss of 3,4-dihydroxyphenylacetic acid (DOPAC), a metabolite of dopamine. The authors hypothesize that these effects are mediated by activation of autophagic/lysosomal pathways. 21 In a mouse model of Huntington disease, rapamycin reduces symptoms of polyglutamine neurotoxicity. Ross/Borchelt mice expressing huntingtin with 82 glutamine repeats treated with rapamycin before the onset of symptoms exhibited reduced numbers of inclusions and improved motor function. 22
Rapamycin may not be altogether beneficial to brain function. mTORC1 plays a key role in synaptic plasticity by modulating protein synthesis. Late-phase synaptic plasticity is known to be attenuated by rapamcyin. Rapamycin has also been shown to disrupt the transition of short-term to long-term memory and interregional memory consolidation in mouse models. 20 However, subtle variation in mTOR activity is likely important, because increased mTORC1 activity also can disrupt memory processing. In fact, in a mouse model of tuberous sclerosis, a disease affecting multiple tissues that causes tuber- or root-shaped growths in the brain, rapamycin treatment rescued memory deficits. Because inhibition of mTOR has been seen to be a possible dietary restriction (DR) mimetic and because DR increases neurogenesis and reduces the rate of aging-dependent brain deterioration and memory loss in rodents, it might be predicted that rapamycin would have similar benefit in humans. Retrospective memory studies on patients treated with rapamcyin for other indications, such as suppression of organ transplantation over long periods of time, might shed light on the usefulness of rapamycin in neurodegeneration, as well as any memory-related side-effects.
Rapamycin and Other Compounds That Target mTORC1 As Antiaging Therapeutics
Inhibition of mTORC1 by rapamycin has substantial beneficial effects in mouse models, including increased longevity and inhibition of engineered neurodegenerative diseases. Intriguing data suggest that rapamycin is also able to suppress HGPS in cultured human cells, making it a potential therapeutic for HGPS as well as neurodegenerative diseases that involve accumulation of toxic protein aggregates. Is there an additional role for mTORC1 inhibitors such as rapamycin to retard aging?
Rapamycin and related compounds carry substantial risk of adverse side effects, which include hypertriglyceridemia (57%), hypercholesterolemia (46%), arthralgia (31%), peripheral edema (58%), fever (34%), pain (33%), anemia (33%), thrombocytopenia (30%), constipation (38%), abdominal pain (36%), diarrhea (35%), nausea (31%), hypertension (49%), and decreased wound healing (
Additional serious potential side effects of rapamycin due to its ability to induce immunosuppression, such as increased rate of infections and cancer, are understudied and probably exaggerated. In fact, there are preliminary clinical data indicating that mTORC1 inhibitors such as rapamycin or everolimus may actually be useful in treating many types of cancers, including relapsed aggressive lymphoma, 25 mantle-cell lymphoma, tumors associated with treatment with cyclosporine, an immunosupressor, 26 neuroendocrine tumors, and metastatic renal carcinoma. 27 Paradoxically, rapamycin actually enhances resistance to some viral infections, such as transplantation-associated late-stage cytomegalovirus. 28 It is possible that the general antiproliferative effects of mTORC1 inhibition on cancer cells outweigh possible reduction in immune surveillance due to reduced Th1 (cell-mediated immunity) and Th17 (tissue lining protection) T cell differentiation and increased maintenance of tolergenic dendritic cells. 29 For infectious diseases, reduced numbers of Th1 and Th17 cells may be counterbalanced by increased antigen presentation due to increased autophagy. 30 It is also entirely possible that rapamycin does not cause more serious side effects because rapamycin-mediated inhibition of mTORC1 is incomplete. 31
The safety profile of rapamycin and related compounds should give pause to its consideration as as a potential antiaging retardant in the absence of potentially life-threatening pathologies. However, there are other compounds that indirectly target mTORC1 with better safety profiles. For example, the antidiabetic drug metformin inhibits mTORC1 by inhibiting RAG guanosine triphosphatases (GTPases) that would otherwise stimulate mTORC1.
32
Metformin and 5-aminoimidazole-4-carboxamide-1-β-
Conclusion
mTORC1 is a promising target for the development of therapeutics for neurodegenerative diseases and HGPS. Rapamycin, an Food and Drug Administration (FDA)-approved drug used to induce immunological tolerance, should be tested for clinical relevance in the treatment of these diseases. However, the substantial set of rapamycin-associated side effects, as well as the lack of aging-specific human data, should caution those who wish to use rapamycin or closely related compounds to slow aging. The use of safer, but perhaps weaker, indirect mTORC1 inhibitors, such as metformin and resveratrol, may prove advantageous. Further study will be useful in ascertaining whether such compounds extend human health or life span.