
Abstract
Lysosomal storage diseases occur due to incomplete metabolic degradation of macromolecules by various hydrolytic enzymes in the lysosome. Despite structural differences, most of the lysosomal enzymes share many common features including a lysosomal targeting motif and phosphotransferase recognition sites. β-Glucuronidase (GUSB) is an important lysosomal enzyme involved in the degradation of glucuronate-containing glycosaminoglycan. The deficiency of GUSB causes mucopolysaccharidosis type VII (MPSVII), leading to lysosomal storage in the brain. GUSB is a well-studied protein for its expression, sequence, structure, and function. The purpose of this review is to summarize our current understanding of sequence, structure, function, and evolution of GUSB and its lysosomal enzyme targeting. Enzyme replacement therapy reported for this protein is also discussed.
Introduction
Deficiencies in activities of lysosomal enzymes that degrade glycosaminoglycans (GAGs) cause an autosomal recessive disease known as MPSVII. GUSB is a 332-kD glycosyl hydrolase that hydrolyzes β-glucuronic acid from the non-reducing termini of GAGs in the lysosome. 7 Deficiency of GUSB causes partial degradation of chondroitin sulfate, dermatan sulfate, and heparan sulfate, which then accumulate in lysosomes of many tissues, 8,9 causing cellular damage and organ dysfunction. This condition is known as mucopolysaccharide storage disease or MPSVII or Sly syndrome. 8,9 It leads to mental retardation, progressive disability, organ dysfunction, dysmorphism, behavioral deficits, and shortened life span. There are 54 different mutations (missense mutations, deletions, nonsense mutations, splice-site mutations) reported in the GUSB gene in the Human Gene Mutation Database that are responsible for MPSVII. Among these mutations, Leu176Phe is one of the most commonly reported mutations in MPSVII patients. 10
Despite its great clinical importance, no concise review of GUSB is available. Therefore, we have compiled all essential information on the sequence, structure, and function of GUSB and its application in enzyme replacement therapy (ERT) in this review.
Biosynthesis and Secretion
The GUSB gene of 25 organisms have been identified and characterized, including in human, 11,12 dog, 13 cat, 14 rat, 15 mouse, 16 and Escherichia coli, 17 and it is classified as a member of the glycoside hydrolase family 2. This gene is expressed in most of the tissues and body fluids. 18 –20 The expression level of GUSB was found to be relatively higher in certain diseases, such as inflammatory joint disease, hepatic disease, and acquired immunodeficiency syndrome (AIDS). 21 The richest source of GUSB is the preputial gland of the female rat where it comprises 7% of the gland's protein. 22
Most of the lysosomal proteins are synthesized as a large precursors on membrane-bound lysosomes, which are subjected to post-translational modifications during or after their transport to the lysosome. 23 Each monomer of GUSB is synthesized on a membrane-bound ribosome and is subsequently translocated to the endoplasmic reticulum (ER) where post-translational modifications occur. The GUSB polypeptide is glycosylated co-translationally and is transferred to microsomal lumen. 24,25 This enzyme undergoes a series of post-translational modifications in higher eukaryotes. 24,25 Li et al. 26 observed that the pro-peptide of GUSB shows sequence similarity with the serpin superfamily (serine proteinase inhibitor) and plays an important role in their compartmentalization.
GUSB is unique among lysosomal enzymes because it shows dual localization to the lysosome and the ER. Egasyn is an enzyme found in the ER and functions as a non-specific carboxyl esterase that metabolizes varieties of compounds. Apart from the esterase activity, egasyn binds to GUSB and sequesters 10–25% of total GUSB in the ER. Although the function of GUSB in the ER is still unclear, a possible role in the hydrolysis of endogenous and xenobiotic glucuronides has been suggested. 27 An eight-residues-long sequence (FGSRPFTF) at the carboxyl terminus of GUSB contains the necessary information to form a complex with egasyn in the mouse. 23 Interestingly, any mutations at the carboxyl terminus of GUSB cause impaired interaction with egasyn. GUSB undergoes several post-translational modifications after its biosynthesis on membrane-bound ribosomes and during its transport to lysosomes. The early processing step includes cleavage of the amino-terminal signal sequence, 24 N-linked glycosylation, 28,29 and the late processing steps, including trimming of carbohydrate chains and proteolytic processing to remove 18 residues from the carboxyl terminus of the protein. 23 One possible function of such a pro-peptide is to serve as a retention signal at ER. The process of carboxy-terminal cleavage and interaction with egasyn will be described later in this review in detail.
Gene Structure and Regulation
The human GUSB gene is located on the long (q) arm of chromosome 7 at position 21.11. 30 The cDNA for mouse, 31 rat, 32 and human 33 of GUSB has been cloned successfully. GUSB is a 21-kb-long gene, comprised of 11 introns and 12 exons of different lengths. 11 Alternate splicing of exon 6 leads to the deletion of 153 bp and gives rise to two different sizes of cDNAs. The human GUSB gene shows close sequence similarity with that of murine, rat and E. coli, 16 indicating that all sequences have evolved from a common ancestor. 34
Shipley et al. 35 have analyzed the upstream sequence required for the expression of the GUSB gene. A 72% G+C content and absence of TATA and CAAT boxes were observed at 200 bp immediately upstream of the translation initiation site. Furthermore, this site contains potential binding sites for the transcription factors such as Spl and activator protein 2 (AP-2). Such unique features are present in all housekeeping genes, which perform essential metabolic functions. Several deletion experiments were performed to determine the minimal length of a 5′ regulatory sequence necessary for GUSB expression. These experiments showed that no more than 200 bp of a 5′ sequence is necessary for the expression of the human GUSB gene. Other regulators of GUSB gene expression were found to be Ca2+ ion concentration and pH. In addition, expression of the GUSB gene can be down-regulated by the calcium ionophore A23187 and the calcium adenosine triphosphatase (ATPase) inhibitor thapsigargin. Kunert-Keil et al. 36 observed the involvement of transcription factor AP-2 binding sites on regulation of GUSB by A23187 through transcriptional mechanisms located in the −354 to +2 fragment of the GUSB promoter. 37 Verapamil (a calcium channel blocker) also decreases the GUSB activity, whereas adeno-associated virus (AAV) enhances the expression of GUSB in the brain. 38
Structure of the Enzyme
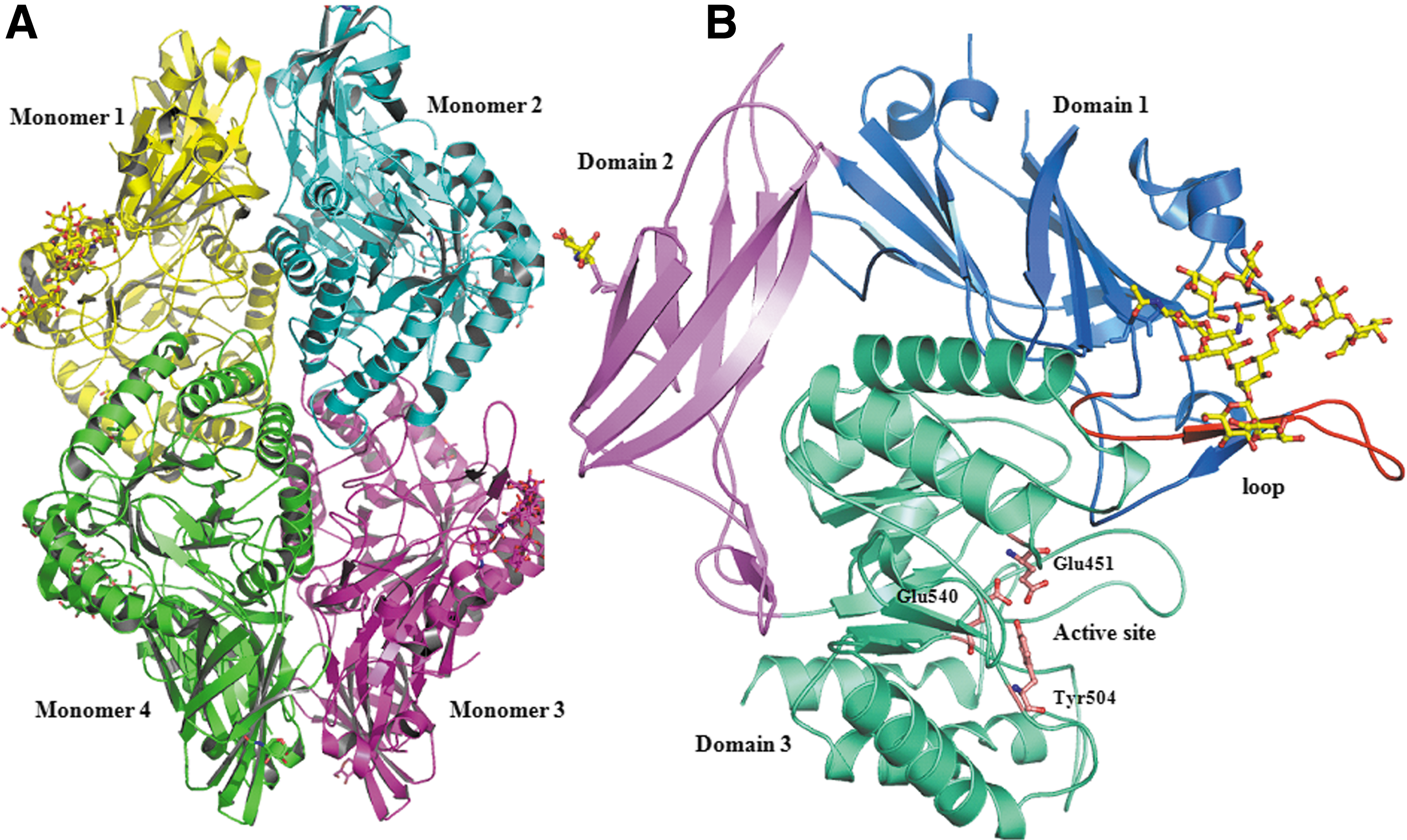
Human GUSB is comprised of 651 amino acid residues (∼78 kD), and contains a 22-residue-long signal peptide and four potential N-linked glycosylation sites. Four subunits form an enzymatically active homotetramer.
39
The crystal structure of human GUSB was determined by Jain et al.,
7
a landmark discovery in the area of lysosomal targeting. This high-resolution crystal structure of GUSB provided a precise mechanism for lysosomal targeting. The GUSB monomer is comprised of three distinct structural domains (Fig. 1). The first domain is a jelly roll shape, which is highly distorted and forma a barrel-like structure along with two β-hairpin insertions. The hairpin loop of the jelly roll motif was considered to be an essential part of GUSB for lysosomal targeting. Therefore, it is also called a lysosomal targeting motif.
7
The second domain is structurally similar to the immunoglobulin constant domains. The third is an α/β or TIM barrel domain motif, which possesses the active site of the enzyme, a characteristic feature of glycosyl hydrolase enzymes. The active site of each monomer is present at the interface of oligomer, and the tetramer complex has four active sites. Multiple sequence alignment of human GUSB with related sequences suggests that active-site residues are highly conserved in all glycosyl hydrolases, revealing their common origin
40,41
(Fig. S1) (Supplementary Data are available at
Three-dimensional structure of GUSB shown in cartoon model. (
Recently, the crystal structure of E. coli GUSB was determined. 42 Despite low sequence similarity (45%) with human GUSB, both proteins share a conserved three-dimensional fold. On comparison of the structure of human GUSB with that of E. coli GUSB, two remarkable differences are observed. In E. coli GUSB, a deletion of four residues in the lysosomal targeting loop (residues 187–200) indicates the role of lysosomal targeting motif in eukaryotic organism. On the other hand, E. coli GUSB contains additional 17 residues in the loop near the active site, which reflects different substrate specificities of the bacterial and human proteins.
Lysosomal Targeting Motif
Sequence analysis and biochemical studies showed that all four potential glycosylation sites (Asn173, Asn272, Asn420, and Asn631) are glycosylated. 43 Glycosylation is important for lysosomal targeting and catalytic activity recognition of protein molecules by the M6PR, which transports lysosomal enzymes to the lysosome. In higher eukaryotes, GUSB undergoes a series of post-translational modifications that direct this enzyme to lysosomes. During transit to the Golgi apparatus, a phosphotransferase catalyzes the transfer of N-acetylglucosamine-1-phosphate from uridine diphosphate (UDP)-GlcNAc to particular mannose residues on the oligosaccharide side chains of lysosomal enzymes. 44,45 This enzyme recognizes a protein conformation shared by lysosomal enzymes that is not present in other secretory proteins. 46 In the Golgi apparatus, a second enzyme removes the terminal N-acetylglucosamine, exposing M6P residues on lysosomal enzymes. 47
The low pH of endosomes degrades the receptor–enzyme complex so that the receptor returns to trans-Golgi via retromer-assisted transport system. 48 Experimental evidence suggests that GUSB is non-glycosylated if expressed in the presence of tunicamycin and exported from the cell without proteolytic processing, supporting the fact that modification of glycans is essential for sorting to final transportation. 49,50 A 46-kD cation-dependent mannose-6-phosphate receptor (CD-MPR) and 300-kD cation-independent mannose-6-phosphate receptor (CI-MPR) are thought to be important for biogenesis and regulation of enzyme secretion. 51,52 Furthermore, defects in transportation cause secretion of the protein. 53 It has been shown that chloroquine-treated enzyme fails to reach the lysosome 54 because it raises the pH of the endosome, and alkaline pH is an unfavorable condition for the enzyme–receptor dissociation.
Structural studies suggest that residues 179–199 of GUSB share sequence and structure homology with the residues 269–283 of human cathepsin D, which were considered to be a “lysosomal targeting motif.” 7 This loop forms a β-hairpin motif and is involved in the phosphotransferase recognition. In addition to this loop, Lys203 was also found to be essential for phosphorylation in cathepsin D. 55 Interestingly, this lysine corresponds to the Lys197 of human GUSB. Alanine-scanning mutagenesis studies showed that Lys203 and Lys293 of cathepsin D are essential residues for phosphorylation and phosphotransferase recognition. 46 A conserved pattern was observed in cathepsin L where Lys54 and Lys99 are essential for the phosphorylation and subsequent phosphotransferase recognition. 56 Mutation of either residue effectively inhibits phosphorylation without causing any remarkable changes in the overall structure of the protein. 57 Interestingly, such lysines are also conserved in the GUSB structure and are suggested to be important for the phosphotransferase recognition. The closeness of the lysosomal targeting motif to the carboxyl terminus of GUSB also suggests the role of carboxy-terminal processing (i.e., the removal of the last two residues) in sorting. 58
GUSB Mutants and Diseases
More than 150 genetic diseases affect the central nervous system (CNS). Of these, more than 30 diseases belong to the lysosomal storage diseases that cause variable degrees of mental retardation to the CNS. 59,60 Most common lysosomal storage diseases occur due to the impaired activity of GUSB, such as Sly Syndrome or MPSVII, which causes mental retardation. 61,62 GUSB shows exoglycosidase activity against various GAGs. Thus, any mutation in the GUSB gene causes malfunctioning of this enzyme, and this is responsible for accumulation of GAGs in the brain, leading to mental retardation. 10 So far, nearly 54 different mutations have been identified in MPSVII patients. 10 These mutations include point mutations, deletions, missense mutations, splice-site mutations, nonsense mutations, and rearrangements. Among the missense mutations, Lue176Phe is considered as a frequent mutation in MPSVII patients and causes a subtle alteration in the structure of GUSB. 63,64
Vervoort et al. 65 identified a pseudo deficiency allele (Asp152Asn) of GUSB and observed a reduced protein stability that causes a remarkable decrease in the enzyme activity. Another mutation reported in the early infantile MPSVII is Tyr626His, which is localized in the last α-helix of the TIM barrel and is in close proximity to two mutations (Ala619Val and Trp627Cys). Such a mutation affects hydrophobic core packing of GUSB. Yamada et al. 66 reported several novel mutations, Pro148Ser, Tyr495Cys, Trp507X, and a 38-bp deletion at position l642–1679 in exon 10 (1642Δ38nt) in MPSVII patients. Among these, the truncating mutation Trp507X produces a more severe MPSVII phenotype. It is interesting to note that no mutation in active-site residues Glu540, Glu451, or Tyr504 is reported in MPSVII patients. Such active-site mutations would possibly produce severe clinical consequences, rendering patients incompatible to survive. 67 A comprehensive analysis of structural changes with respect to mutation sites have already been reported earlier. 10 Interestingly, approximately 90% are point mutations among all mutations reported in the GUSB gene of MPSVII patients.
Enzyme Replacement Therapy
ERT prevents the accumulation of lysosomal storage in MPSVII patients. 62 However, if untreated, deficiency in GUSB activity leads to the progressive accumulation of partially degraded GAGs in many tissues. 68 Sands et al. 69 showed that multiple injections of murine GUSB containing ∼70% phosphorylated enzyme prevent the accumulation of lysosomal storage in many tissues, with the exception of the brain. 70 Phosphorylation of GUSB is also an essential parameter for the clearance of GAGs. Sands et al. 71 further showed that both phosphorylated and non-phosphorylated forms of GUSB contribute to reduction in lysosomal storage. However, highly phosphorylated GUSB reduces storage in a wider range of cell types and results in a more complete clinical response than that seen using completely non-phosphorylated enzyme. 71 Wolfe et al. 72 suggest that retroviral vector–mediated transfer of the GUSB gene to mutant stem cells resulted in a long-term expression of low levels of GUSB. Interestingly, such transfer partially reduces lysosomal storage in liver and spleen and corrects the disease.
Because chemotherapy has severe side effects, glucuronide-based pro-drugs are widely used as anti-cancer agents. Hence, GUSB plays important role in the treatment of pancreatic carcinoma because it releases the active moiety of glucuronide pro-drugs within or near the tumor. GUSB has been used for the treatment of cancer through antibody-directed enzyme pro-drug therapy (ADEPT). Because GUSB acts specifically on the glucuronides, pro-drug systems of glucuronides derivative of daunomycin and adriamycin have been studied. 73 In this approach, the patient is injected with a fusion protein containing a GUSB moiety and humanized anti-carcinoembryonic antigen (CEA) monoclonal antibody. Administration of this fusion protein can be used in treatment with a glucuronide pro-drug such as N-(4-glucuronyl-3-nitrobenzyloxycarbonyl)doxorubicin, a glucuronide pro-drug of doxorubicin. This is a non-toxic derivative of doxorubicin in which a glucuronide acid moiety is coupled to the anthracycline molecule via synthetic spacer. Such a unique pro-drug–based system is comprised of a functionally active fusion protein containing a humanized tumor-selective binding portion and GUSB, which has potential for selective tumor therapy. The main purpose of using GUSB in ADEPT is to avoid the development of an immunological response. 74
ERT has been successfully used for the treatment of many lysosomal storage diseases. 75 –77 Recently, a monoclonal antibody–based GUSB fusion protein was characterized to assess the in vivo pharmacokinetics, brain uptake, and enzyme activity in rhesus monkeys. 78 In this study, a Chinese hamster ovary–derived human insulin receptor–iduronate-2-sulfatase fusion protein was shown to retain high enzyme activity and high-affinity binding to the human insulin receptor. However, correction of brain storage is limited by the inability of infused enzyme to cross the blood–brain barrier. Grubb et al. 79 chemically inactivated the terminal sugars of GUSB by treatment with sodium metaperiodate, followed by borohydride reduction (PerT-GUS). 80 In the murine MPSVII model, long-circulating periodate-treated GUSB was more effective at clearing storage from the CNS than the native GUSB. It was shown that PerT-GUS retains nearly 75% of the enzyme activity. Furthermore, such modified enzyme showed increased sensitivity toward heat inactivation. In addition, kinetic properties of both the native and modified GUSB were similar. 79 Interestingly, the modified GUSB had a longer half-life in circulation and was more effective in clearing neuronal storage than the native GUSB.
MPSVII, due to the deficiency of GUSB, often leads to an increase in the levels of other lysosomal enzymes such as α-galactosidase and β-hexosaminidase. 9,81 This process is known as secondary elevation and is considered a useful biomarker for the effectiveness of ERT in reducing lysosomal storage. 69,82,83 Treatment of MPSVII mice either with the native GUSB or PerT-GUSB showed a significant reduction in secondary elevation of α-galactosidase enzyme in the brain after 12 weeks. Furthermore, PerT-GUSB treatment causes greater reduction in α-galactosidase activity in the brain as compared to the native GUSB in the brain. Moreover, in the liver, both PerT-GUSB and native GUSB showed complete correction of secondary elevations of α-galactosidase. This observation suggests that delivery of both enzymes to liver exceeded the threshold required for complete correction of secondary elevation. On the other hand, in the brain, PerT-GUSB treatment produced a remarkable decrease in β-hexosaminidase activity in the brain. No significant decrease in β-hexosaminidase levels was observed by the native GUSB in the brain. However, in liver, both enzymes completely corrected β-hexosaminidase levels. All of these findings suggest that PerT-GUS is superior to the native enzyme in crossing the blood–brain barrier, correcting neuronal storage, and significantly reducing secondary elevation of α-galactosidase and β-hexosaminidase. 84 Furthermore, infusions of PerT-GUSB versus native GUSB showed improved delivery of enzyme to the brain and corrected secondary elevations of other lysosomal enzymes and a significant improvement in histopathology as well. PerT-GUSB was found to be superior to native GUSB in all the three categories, thus such features of PerT-GUSB may be considered as biomarker for correction of neuronal storage. 85 Finally, these observations provide additional evidence that chemically modified long-circulating enzyme escapes carbohydrate-mediated clearance and may offer advantages in MPSVII treatment. 84
Recently, Rowan et al. 86 extended the application of PerT-GUSB over the native GUSB for the treatment of skeletal diseases. They performed analysis of bone pathology of MPSVII mice using micro-computed tomography (CT) to observe the three-dimensional structure of bone, an objective measure of bone mineral density. To address the question whether PerT-GUSB treatment can improve bone pathology and clear skeletal GAG storage, they compared the skeletal response of MPSVII mice after 12 weeks of treatment with PerT-GUSB or native GUSB. Histopathological analysis showed a significant reduction in storage material and a more organized growth plate in PerT-GUSB–treated mice in comparison to the native GUSB. Interestingly, thickness of the articular cartilage in GUSB and PerT-GUSB–treated mice was similar. However, the growth plates in native GUSB-treated MPSVII mice showed an increase in thickness as compared with that observed with PerT-GUSB. Moreover, micro-CT and X-ray studies showed a greater reduction in bone mineral density and reduced femur thickness, respectively, with PerT-GUSB treatment. Finally, they also showed that PerT-GUSB treatment also corrects CNS storage in MPSVII mice and significantly prevents skeletal pathology as compared to the native GUSB enzyme. In conclusion, PerT-GUSB showed great impact on the treatment of bone lesions as well as CNS storage. This ERT can be used for the treatment of other lysosomal storage diseases and brain lesions.
Gene therapy for MPS has been reviewed extensively by Ponder and Haskins. 3 Gene therapy is the successful transferring of a gene into an animal that either encodes active protein or corrects mutations in the endogenous gene that results in long-term expression in the deficient animal. 87,88 Interestingly, intravenous injection of an AAV containing single-stranded DNA has been used to correct a single-base deletion in the endogenous mutant of the GUSB gene of MPSVII mice. 89 This process does not bear any fruitful results toward disease correction, but such an approach presumably controls gene regulation. On the other hand, when very high dose of GUSB was injected intravenously into the adult MPSVII mice, 2.5% of GUSB activity was retained in the brain. 90 Furthermore, lysosomal storage in the brain can be reduced with systemic or intra-hepatic gene therapy without vector copies in the brain. 91 –93 These remarkable findings support the fact that some enzymes in blood can cross the blood–brain barrier if their levels are sufficiently high.
The intravenous injection of AAV-containing the GUSB gene in newborn MPSVII mice showed a widespread distribution of vector throughout the body, including the brain. 94 This suggests that AAV is a promising tool for direct gene transfer into the brain along with the maintenance of gene expression for longer time in this organ. 95,96 Injection of recombinant adenovirus to the lateral ventricles of MPSVII mice showed a 30% increase in GUSB activity in ependymal cells and choroid, whereas low activity was observed in the brain parenchyma associated with vessels and in meninges. 97 Moreover, the route of vector administration plays significant role in the extent and distribution of GUSB activity. 98 Serguera et al. 99 used primary adult human astrocytes as the vehicle for GUSB delivery in the brain and showed that intravenously injected acidic amino acid–tagged enzyme had five times more prolonged blood clearance compared to the native enzyme. The tagged enzyme was delivered effectively to bone, bone marrow, and brain in MPSVII mice and was active in reversing the storage pathology. 100 Because the acidic amino acid-tagged enzyme is cleared at a slow rate after infusion, this enzyme is able to reach cells outside hepatocytes and the reticulo-endothelial system. This strategy helps in targeting GUSB to specific cell-surface receptors and its transport to the lysosome by receptor-mediated endocytosis. Several novel approaches for gene transfer are designed and in progress to increase the systemic levels of GUSB in the MPSVII mouse. 101
Brooks et al. 102 showed a bi-hemispheric correction of the characteristic cellular pathology of GUSB-deficient MPSVII mice when injected with recombinant feline immunodeficiency virus expressing GUSB. However, after injection of virus-based vectors expressing GUSB into the brains of MPSVII mice, a dramatic recovery of behavioral function, such as spatial learning and memory, was observed, suggesting that ERT can have effects beyond restoration of GUSB activity in the lysosome and can impart improvements in plasticity and spatial learning. Sly and Vogler 103 commented that given the rapidly expanding number of animal models of lysosomal storage diseases with CNS involvement, and the generality of the biology of lysosomal enzyme transport, studies like that of Brooks et al. 102 are likely to be replicated in other animal models. They suggested that if the results are as promising as those presented for murine MPSVII, the Brooks et al. 102 study will likely be viewed as a landmark that took the field well beyond the blood–brain barrier.
GUSB Functions
GUSB is a housekeeping enzyme expressed in most tissues and is actively involved in degradation of proteoglycans in lysosomes. It plays a significant role in the degradation of dermatan and keratan sulfates by catalyzing the fifth step of degradation of GAGs. GUSB is also involved in cation binding and inter-conversions of various metabolites such as pentose, glucuronate, chlorophyll, porphyrin, starch, and sucrose. GUSB plays essential role in remodeling of the extracellular matrix components in both physiological and inflammatory states. In addition, GUSB incorporates β‐glucuronides to steroid hormones 104 and helps in recirculation of vitamin D, thyroid hormone, bilirubin, and estrogen. 105 –107 GUSB plays a significant role in the release of active or inactive compound from drug glucuronides and therefore modifies disposition and action of pro-drugs. 108 Non-steroidal anti-inflammatory drugs are carboxylic acid–containing compounds that are conjugated in the liver to acyl glucuronides and excreted into bile. The glucuronic acid moiety is cleaved off the glycone due to change in local pH in the presence of GUSB. 109
GUSB helps to de-conjugate various potential toxins, hormones, and various drugs in the body.
110,111
It is also involved in hydrolysis of glucuronides of endo- and xeno-biotics in human due of its hydrolyzing activity against glucuronide conjugate.
112
GUSB plays a significant role in the cleavage of endogenous compounds to increase the activity in bile in pathogenesis of pigment cholelithiasis. Whiting et al.
113
demonstrated that microsomal GUSB influences the rate of biliary excretion of bilirubin-IXa glucuronides. Separation of calcium bilirubinate from the bile is also catalyzed by GUSB. GUSB hydrolyzes bilirubin glucuronide into free bilirubin and glucuronic acid. Calcium of bile combines at the carboxyl radical of liberated bilirubin to form calcium bilirubinate.
114
The function of GUSB has also been reported in several intermediary metabolic pathways.
113,115,116
It is involved in the biosynthetic path of
GUSB is expressed in various plants (skullcap, rye, rhubarb, duckweed, sugar beet, and tobacco).
117,118
GUSB from Scutellaria baicalensis has shown high-level activity against flavone 7-O-β-
GUSB Enzyme Mechanisms
GUSB is a well-studied enzyme, and its assay procedures were described nearly 60 years ago.
123,124
The kinetics of GUSB have been well described for chick embryo, human placenta, and the rat preputial gland using the substrate, 4-methylumbelliferyl-β-
The kinetically active form of GUSB is a homo-tetramer. 7 However, it shows appreciable activity in the dimer form. 39 Matsuura et al. 127 determined the rate constants of the monomer to tetramer assembly and showed that GUSB has two rate-limiting steps—monomer to dimer assembly and dimer to tetramer assembly. Crystal structure analysis 7 suggests that active-site residues Glu451, Glu540, and Tyr504 are positioned at an optimum distance to perform nucleophile attack and acid–base catalysis. Wong et al. 128 proposed a precise two-step mechanism of GUSB activity. The first step is nucleophilic attack by one of the carboxylates on the sugar anomeric center. In the second step, the intermediate is hydrolyzed by general base-catalyzed attack of water at the anomeric center, resulting in the cleavage of glycosidic bond with net retention of the anomeric configuration. Formation and hydrolysis of the glycosyl-enzyme intermediate proceed via transition states with a substantial oxocarbenium ion character.
Biochemical and structural analyses suggest that active-site residues are highly conserved in the E. coli β-galactosidase. 129 Four critical residues are part of the active-site loop and are essential to provide substrate specificity to GUSB. These residues are Tyr509, Ser557, Asn566, and Lys568. 130 The specificity of these residues was predicted by a molecular docking approach. It was observed that β-glucuronic acid docks in the active site of GUSB without altering the enzyme structure. Interestingly, eight intramolecular hydrogen bonds are formed between the substrate and seven residues of GUSB, including Tyr509, Ser557, Asn566, and Lys568. Furthermore, site-directed mutagenesis studies have confirmed that mutants with Tyr509Ala, Ser557Pro, Asn566Ser, and Lys568Gln have lost their substrate specificity significantly. This observation was further supported by mutations in loop eight of GUSB, which was believed to be the most divergent part of the active site and is responsible for the C5 specificity. Finally, Geddie et al. 131 proposed a model that showed GUSB and β-xyloside substrates differ only in their C5 substituent, so it may be possible that single amino acid replacement in GUSB would be sufficient to convert it into a xylosidase.
Functional Networking Partners
GUSB interacts with large number of proteins to perform several biological tasks (Fig. 2). To know the functional protein association networks of GUSB with various proteins, we performed STRING
132
analysis and listed the top 20 proteins that showed close interaction with GUSB (Table 1). The interaction analysis of STRING is based on both experimental and theoretical interaction information. A list of interactions in STRING is provided with a confidence score and related information (
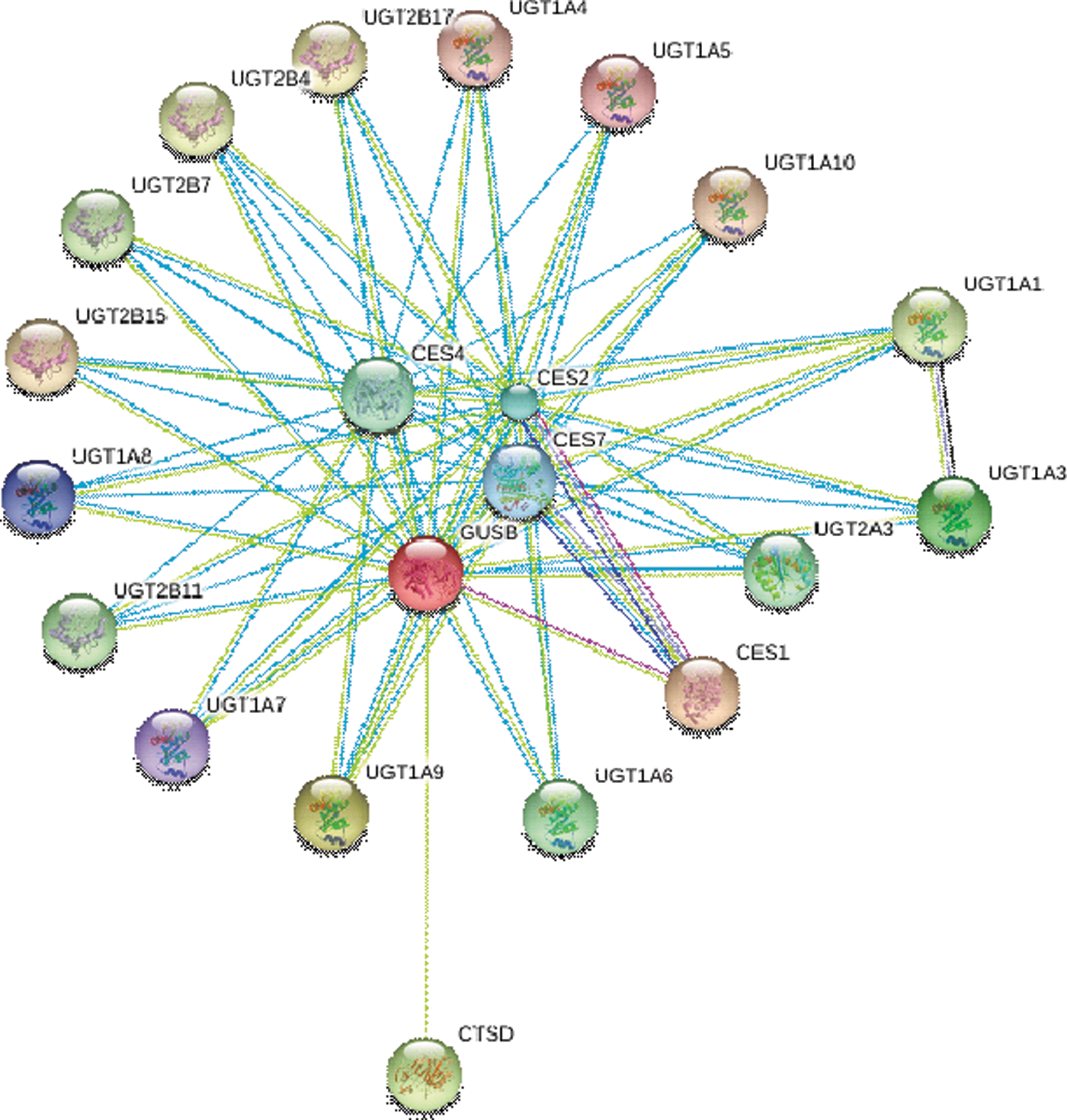
Functional protein interaction network of GUSB calculated by the online server STRING (
E, experimental; T, text mining; D, databases.
Interestingly, most members of UDP glucuronosyltransferase 1 (UGT) family show appreciable interaction with GUSB. UGT is involved in detoxification of endo- and xeno-biotics, including many drugs. 135 The intralumenal orientation of the active site of UGTs in the ER membranes binds a large number of molecules, including UDP-glucuronic acid and glucuronides. 136 In all lysosomal enzymes, carbohydrates are modified to form a phosphorylated recognition marker. The mannose residue of lysosomal enzymes, especially GUSB, is phosphorylated at C-6 by UDP-N-acetylglucosamine with the help of the enzyme UGT. Such processing of the oligosaccharides in lysosomal enzymes makes them recognizable by M6PR and directs translocation of lysosomal enzymes from Golgi apparatus to the lysosomes. 137
Conclusions
GUSB is a glycosyl hydrolase that catalyzes cleavage of β-
Footnotes
Acknowledgments
M.I.H. is thankful to University Grants Commission (UGC) and Indian Council of Medical Research for financial assistance. The authors are grateful to Indo-US Science and Technology Forum for providing funds to establish an active collaboration between M.I.H. and W.S.S. F.A. is thankful to the Council of Scientific and Industrial Research (CSIR) for financial support. H.N. expresses her thanks to Jamia Millia Islamia for a fellowship.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.