
Abstract
The dysfunctional changes of aging are generally believed to be irreversible due to the accumulation of molecular and cellular damage within an organism's somatic cells and tissues. However, the importance of potentially reversible cell signaling and epigenetic changes in causing dysfunction has not been thoroughly investigated. Striking evidence that increased oxidative stress associated with hematopoietic stem cells (HSCs) from aging mice causes dysfunction has been reported. Forced expression of SIRT3, which activates the reactive oxygen species (ROS) scavenger superoxide dismutase 2 (SOD2) by de-acetylation to reduce oxidative stress, functionally rejuvenates mouse HSCs. These data, combined with numerous other reports, suggest that ROS act as a signal transducer to play a critical regulatory role in HSCs and at least in some other stem cells. It is likely that ectopic expression of SIRT3 restores homeostasis in gene expression networks sensitive to oxidative stress. This result was surprising because age-associated damage from impaired DNA repair had been thought to be irreversible in old HSCs. The effect of up-regulated SIRT3 in HSCs is one of first examples in which intrinsic cellular aging, not apparently associated with changes in the micro-environment, was reversed. However, the stability of rejuvenation in the absence of continued supplemental SIRT3 expression was not investigated. These data are consistent with a hypothesis that potentially reversible processes, such as aberrant signaling and epigenetic drift, are relevant to cellular aging. If true, rejuvenation of at least some aged cells may be simpler than generally appreciated.
Introduction
One of the most potentially insightful and relevant debates in aging research is whether oxidative stress/redox/reactive oxygen species (ROS) contribute to aging by causing the accumulation of damage in the mitochondria in a self-perpetuating positive feedback loop, or alternatively by sending dysregulated homeostatic signals (noise). Although many studies have reported a connection between ROS and molecular damage, evidence is accumulating in model organisms, such as nematodes, fruit flies, and even mice, that ROS can alter the rate of aging through signal transduction mechanisms 9 and probably plays an important role in control of cell proliferation, 10 differentiation, 11 –13 metabolism, 14 and death. 15 For example, endogenous ROS are necessary for proliferation of neural stem and progenitor cells 16 and for tail regeneration in Xenopus tadpoles. 17 One relatively unexplored possibility is that ROS signal by modifying the gene networks and the epigenome by directly acting on redox-sensitive chromatin remodeling proteins and transcription factors, which might have profound implications for explaining age-correlated epigenetic changes. 18
If ROS signaling is as important to homeostasis as recent reports hint, then understanding how ROS levels are controlled is critical to untangling its role in aging. SIRT3, a mammalian member of the sirtuin family of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases, deacylases, and adenosine diphosphate (ADP)-ribosyltransferases, 19 is among the more intriguing candidates for a regulator of ROS. 20 Sirtuins have been implicated in aging since it was found that Sir2, a canonical sirtuin, was necessary for increased longevity associated with caloric restriction (CR) in yeast and that over-expression of Sir2 extended life span in a variety of invertebrates. However, the connection between longevity and Sir2 over-expression in worms and flies is controversial, 21 and only over-expression of SIRT6 has been found to increase longevity in a mammalian model system and then only male mice. 22 Similarly, the role of resveratrol as a sirtuin activator has been debated, although the latest evidence suggests that resveratrol acts directly as an allosteric activator of SIRT1. 23 Nevertheless, sirtuins do play a role in metabolic regulation and of the seven mammalian sirtuins; three—SIRT3, SIRT4, and SIRT5—are mostly localized to the mitochondria, where SIRT3 and SIRT5 help regulate mitochondrial enzyme activities and metabolic adaptation 24 by altering protein acetylation. SIRT3 expression, along with SIRT1 and SIRT6, increases during CR. 25 –27 SIRT3 affects numerous proteins in electron transport, 20 is required for the activity of the mitochondrial biogenesis master regulator peroxisome proliferator-activated receptor-γ co-activator (PPARγ) in brown adipocytes and skeletal muscle, 28,29 modulates cell death in response to injury or genotoxic damage, 30,31 is required for CR to promote long-term survival of inner ear cells involved in hearing that are normally lost during aging, 26 and prevents cardiac hypertrophy associated with aging in mice. 32,33 SIRT3 controls ROS levels by stimulating ROS scavengers superoxide dismutase 2 (SOD2) 34,35 and possibly catalase. 35 During CR, SIRT3 directly de-acetylates SOD2 to increase superoxide scavenging, 36 while extra-mitochondrial SIRT3 also can de-acetylate the FoxO3A transcription factor, which then trans-locates to the nucleus to increase SOD2 and catalase transcription 35 (but this was not confirmed in other work, see ref. 36). Furthermore, SIRT3 indirectly increases the levels of reduced glutathione, another important cellular anti-oxidant, by increasing isocitrate dehydrogenase 2 activity, which increases nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) levels, which in turn stimulates glutathione reductase. 26
Increased SIRT3 Expression Rejuvenates Aging Hematopoietic Stem Cells
ROS levels increase in hematopoietic stem cells (HSCs) with age, resulting in increased apoptosis and proliferation, which eventually results in diminished self-renewal capacity and differentiation. A positive feedback loop in which ROS result in mitochondrial molecular damage, which in turn causes increased ROS, has been hypothesized to cause irreversible stem cell quiescence, senescence, and death. Aging of HSCs in particular has been thought to be irreversible due to cell-intrinsic, irreversible changes in DNA repair. 4,37,38 In an important paper, Brown et al. 39 show that forced over-expression of SIRT3 in HSCs from old mice restores HSC regenerative capacity. We believe that the simplest explanation is that SIRT3 attenuates dysfunctional mitochondrial ROS signaling, which acts as a source of epigenetic noise to disrupt gene regulatory networks sensitive to redox and oxidative stress, which in turn perturbs networks that control HSC function.
To dissect the role of ROS and SIRT3 in HSC function, Brown and colleagues first tested the hypothesis that loss of SIRT3 increases ROS and reduces function in HSCs. In young mice, SIRT3 is expressed at 3000-fold greater levels in HSCs than in differentiated blood cells, and the authors observed that SIRT3 expression decreases in old animals, so the authors expected that the HSCs of SIRT3 knockout mice (SIRT3-KO) might exhibit premature aging. However, they found no differences in the numbers of HSCs (Lin−, c-Kit+, Sca1+, CD150+, CD48−) or hematopoietic stem/progenitor cells (HSPCs)((Lin−, c-Kit+, Sca1+) from SIRT3-KO mice. (HSCs are a subset of hematopoietic stem and progenitor cells [HSPCs]; Brown and co-workers generally observed similar results for both populations.) HSCs from young SIRT3-KO animals show normal ROS levels and are equally adept as wild type (WT) mice at reconstituting the hematopoietic compartment of lethally irradiated mice. No difference in the number of blood cell types derived from the WT and SIRT3-KO HSCs was observed. 39
ROS levels in HSCs are known to increase significantly as animals age 40 and after three cycles of serial transplantation. HSCs derived from SIRT3-KO mice demonstrated a 50% reduction in self-renewal compared to WT mice after three serial transplantations. Consistent with this observation, HSC compartments were 50% smaller in aged 18- to 24-month-old SIRT3-KO mice compared to WT, and the reconstitution efficiency of aged HSCs was 30% less in for SIRT3-KO cells. The authors concluded that SIRT3 is necessary to maintain HSC pool size and function with increasing age. 39 However, there is another important conclusion that the authors do not reach: Because SIRT3 is not necessary to prevent high ROS or dysfunction in young animals, loss of SIRT3 is not the only defect in aging HSCs causing increased ROS. The critical question that remains is the cause of the increased ROS levels with age. The question remains unanswered.
Brown et al. studied whether SIRT3-KO affects the micro-environment/niche by transplanting HSCs from WT donors into SIRT3-KO mice. If the absence of SIRT3 altered the stem cell micro-environment, then less self-renewal, reconstitution, and differentiation would be observed in SIRT3-KO mice. No difference in HSC function was seen between irradiated SIRT3-KO and WT mice, suggesting that SIRT3 does not act extrinsically to affect the micro-environment. 39
SIRT3 is known to increase activity of anti-oxidant enzymes such as SOD2 to reduce ROS levels. Although knockout of SIRT3 did not alter ROS levels in HSPCs of young animals, SIRT3-KO did increase ROS in aged HSPCs and under transplant stress. To assess whether the (decreased) presence of SIRT3 still protects old HSPCs from elevated ROS through SOD2, SOD2 expression and activity were compared between old WT and SIRT3-KO HSPCs. HSPCs had equivalent levels of SOD2 mRNAs, but WT mice had 50% more SOD2 activity, whereas SIRT3-KO mice had 40% more dysfunctional mitochondria as assessed by Mitotracker fluorescent dyes. The authors conclude that SIRT3 regulates mitochondrial homeostasis and integrity in HSPCs. 39 To further clarify the role of SOD2, old HSCs were infected with lentiviruses that ectopically expressed either WT SOD or with SOD2 carrying K53R and K89R mutations, which prevent acetylation. The mutant is as catalytically active as de-acetylated WT SOD2. Interestingly, only forced expression of mutant SOD2 increased colony-forming activity (by 75%); WT SOD2 had no effect. This data is consistent with the hypothesis that SIRT3 acts at least in part by de-acetylating SOD2, which then reduces ROS by dismutation of superoxide ions. Normative ROS levels then de-activate oxidative stress–sensitive gene networks that interfere with function. One implication of this result is that old HSCs have high levels of mitochondrial protein acetylation, which may be caused by lower SIRT3 expression.
Oxidative stress disrupts the normal quiescent state that protects HSCs from over-proliferation and subsequent senescence to maintain self-renewal capacity.
41
Increased cell cycle activity and decreased survival were observed in aged HSPCs, suggesting that possible disruption of function was due to oxidative stress/ROS. This led the authors to test whether reduction of ROS by other means could rescue old SIRT3-KO cells. Treatment with the powerful anti-oxidant N-acetyl-
Most importantly, experiments employing forced over-expression of SIRT3 by transduction with SIRT3 lentiviral vectors similarly reduced ROS and restored regenerative capacity of aged and SIRT3-KO HSCs. 39 This is one of first demonstrations that defects associated with intrinsic aging at the cellular level can be reversed in stem cells and that aging cells can be rejuvenated.
Possible Mechanisms for SIRT3 Rejuvenation of HSCs
There are two important questions: (1) How does ectopic expression of SIRT3 rejuvenate HSCs? and (2) How throughly does SIRT3 rejuvenate HSCs?
SIRT3 is clearly not necessary to maintain normative ROS levels and HSC function in young animals; nevertheless, it can rescue HSC function and restore ROS levels in old animals. The authors provide strong evidence that SIRT3 reduces high ROS levels by de-acetylating SOD2 to stimulate removal of superoxide radicals. The resulting absence of high ROS levels acts as signal to terminate oxidative-stress responses in gene networks that inhibit HSC function. One possible mechanism by which ROS may destabilize HSC function is though activation of Nrf2, a transcription factor that acts as an oxidative stress sensor. In the presence of ROS, Nrf2 trans-locates from the cytoplasm to the nucleus to initiate transcription of a protective response at genes regulated by anti-oxidant response elements. In a recent report, Nrf2 was shown to regulate HSC function and renewal. 42 We speculate that chronic Nrf2 activation may suppress HSC function, but that hypothesis needs to be investigated.
To answer how thoroughly SIRT3 rejuvenates HSCs requires defining what is necessary for complete rejuvenation. Is restoration of HSC function sufficient or does full rejuvenation require removing additional damaged biomolecules or resetting the cell's epigenetic state such that ROS levels will not immediately return to high levels in the absence of supplemental SIRT3? In other words, it is necessary to understand why ROS levels increased with aging in the first place? One possibility is that increased ROS levels result from accumulating damage that also somehow causes lowered SIRT3, as the authors suggest. Another possibility is that the homeostasis of ROS signaling is disrupted by stochastic fluctuation in signal transduction or perhaps epigenetic drift, such as seen in the human methylome. 43,44 In the latter case, a close examination of possible connections between the genes whose DNA methylation state best correlates with age and SIRT3 is warranted. A unifying possibility might be that the use of ROS for cell signaling is itself the source of gene regulatory and epigenetic perturbations, mediated by redox- and oxidative stress-sensitive transcription factors and epigenomic regulators.
One way to gain insight into the extent of rejuvenation is to assess whether the effects of chronic oxidative stress are completely removed by the initial treatment. If so, switching off the ectopically expressed SIRT3 or terminating treatment with NAC should result in a gradual return to high ROS and the dysfunctional state over time. If the underlying defect was only masked by by-passing upstream permanent damage or epigenetic information loss, then removing SIRT3 or NAC should result in an immediate return to high ROS and the dysfunctional state.
General Implications for Rejuvenation of Aged Cells
Work on HSCs and satellite skeletal muscle stem cells, for example, suggests that rejuvenation of some cell types is possible. For comprehensive rejuvenation, the true extent of irreversible molecular damage, potentially reversible epigenetic changes, and reversible disruption of cell signaling/proper cellular milieu must be assessed, so that appropriate techniques to overcome this damage can be developed and applied. For example, the SENS Foundation has cataloged a number of ways in which molecular, cellular, and extra-cellular damage can be removed and repaired. Obviously, the more plastic the molecular basis of the dysfunction is, potentially the easier it can be addressed. For example, DNA mutations that remain after endogenous DNA repair are essentially irreversible and could only be corrected by the development of safe genome engineering. On the other hand, disruption of gene regulatory networks by aberrant signaling may be treatable by drugs, similar to the effects of NAC on ROS in HSCs in vivo. Epigenetic alterations that stably alter differentiation state, such as those seen in epigenetic drift, lie in a middle ground. It is unclear whether such cells would first require full reprogramming to a pluripotent state followed by re-differentiation, which is completely impractical in vivo, or could be directly stimulated to re-differentiate to their own youthful cell state using methods similar to those used to trans-differentiate cells.
Medical Implications
The loss of SIRT3 expression with age or stress demonstrated in HSCs may be more ubiquitous than reported in the work of Brown et al. It will be of great interest to assess mitochondrial function, ROS, and SIRT3 levels in other tissues and stem cells to see how widespread this effect is. It will be especially important to determine if SIRT3 is down-regulated with age and whether enforced SIRT3 up-regulation can rescue function in human HSCs or other human stem cells.
Given these intriguing results, are there any known ways to raise SIRT3 expression levels? Three ways have been reported (Fig. 1) —exercise, 45 CR, 46 and treatment with angiotensin (AT1) antagonists, such as losartan. 47 Interestingly, all of these have been reported to extend longevity in at least some animal models.
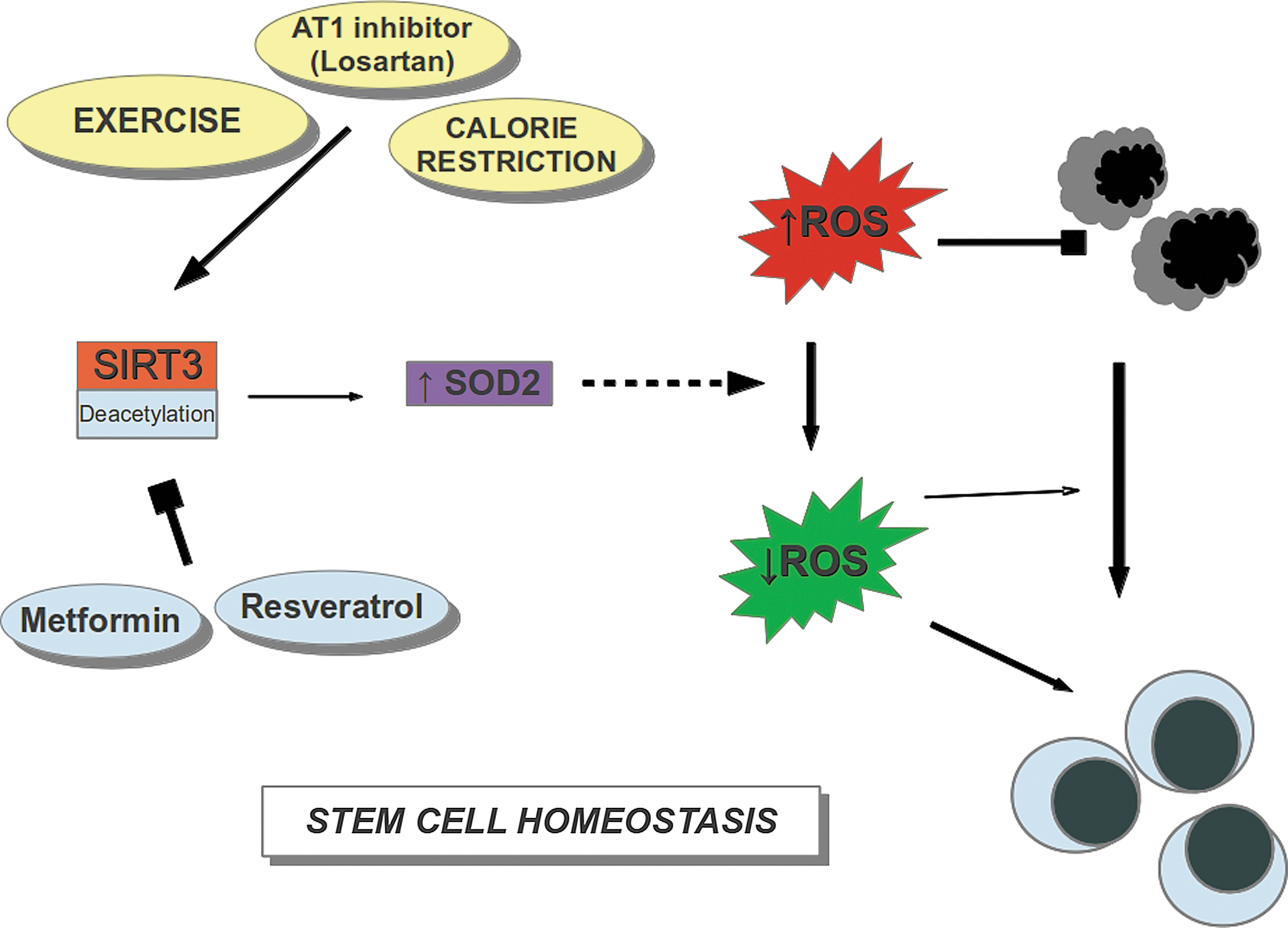
SIRT3 rescues hematopoietic stem cell (HSC) function by reducing reactive oxygen species (ROS). HSC function is inhibited by high ROS levels in old cells (grey and black). SIRT3 stimulates antioxidant superoxide dismutase 2 (SOD2) activity by deacetylation. SOD2 reduces ROS levels, restoring HSC function (cyan cells). Exercise, calorie restriction (CR), and angiotensin (AT1) inhibitors increase SIRT3 expression. Resveratrol inhibits SIRT3 activity and metformin inhibits SIRT3 expression. (Color images available at
Just as important is the observation that some compounds thought to extend longevity or to be of benefit in slowing some aging-related processes actually may inhibit SIRT3 expression or activity (Fig. 1). Among these are resveratrol (and some of its metabolites), which at high doses interact with and inhibit SIRT3 enzyme activity, 48 and metformin, which inhibits expression of SIRT3. 49 Perhaps this is one of the reasons why life span extension in mammals is modest at best with metformin and absent in animals fed standard diets supplemented with resveratrol.
Conclusion
The effects of aging are generally thought to be irreversible, due to the accumulation of molecular damage. However, Brown and colleagues show that even presumed intrinsic aging in stem cells may be reversible. Ectopic expression of SIRT3, expression of a constitutively de-acetylated SOD2, and NAC all lower ROS levels, such that HSC function is restored, possibly by terminating oxidative stress–mediated disruption of gene networks involved in regulating HSC function. Whether the HSCs are stably rejuvenated by SIRT3 remains unclear, in terms of whether they retain residual damage or hidden epigenetic changes. The reversibility of aging-associated dysfunction in other somatic cells and stem cells may depend upon the dynamism or plasticity of the underlying molecular changes. If these results are found to hold in humans, the use of AT1 antagonists might have benefit for older people who have reduced SIRT3 expression that leads to immune dysfunction.
Footnotes
Author Disclosure Statement
No competing financial interests exist.