
Abstract
Recent breakthroughs have provided notable insights into both the pathogenesis and therapeutic strategies for age-related hearing loss (ARHL). Simultaneously, these breakthroughs enhance our knowledge about this neurodegenerative disease and raise the question of whether the disorder is preventable or even treatable. Discoveries relating to ARHL have revealed a unique link between ARHL and the underlying pathologies. Therefore, we need to better understand the pathogenesis or the mechanism of ARHL and learn how to take full advantage of various therapeutic strategies to prevent the progression of ARHL.
Introduction
A
Although the potential mechanisms underlying ARHL have not been established, the age-related decline in acuity is generally attributed to at least two factors: (1) Progressive peripheral degeneration involving the degeneration of the stria vascularis, 5 hair cells, spiral ganglion neurons (SGNs), 6 and fibrocytes, 7 leading to the disability of the sound afferent, and (2) a central degeneration that involves neuron degeneration in the primary auditory cortex (AI), 8,9 thalamus, 10 synapses in the hippocampal CA3 region 11 and the dorsal cochlear nucleus (DCN) 12 that results in various degrees of the central nervous system (CNS) executive dysfunction to various degrees in many patients with this disorder. The symptoms and signs emerge when these two factors are present. Mouse models are used to mimic different forms of human ARHL. Although the cochlear regions of a given type of pathology in animals may be different from humans, they exhibit similar degeneration of the organ of corti and SGNs, except for the stria vascularis. 13 Schuknecht 14 reported that approximately 30% of the patients with ARHL exhibit degeneration of the stria vascularis. Thus, animal models are appropriate for studying human ARHL.
The age of clinical onset is variable, but ARHL usually begins after 60 years. Neither the pathogenesis nor the symptoms of the disease have a remarkable onset; however, the disease progresses relentlessly. Recent discoveries related to ARHL have revealed a unique link between ARHL and the underlying pathologies. Therefore, we need to better understand the pathogenesis or the mechanism of ARHL and learn how to take full advantage of various therapeutic strategies to prevent the progression of ARHL. Accumulating evidence 15,16 from genetic and electrophysiological studies postulates that reactive oxygen species (ROS)-related mitochondrial (mtDNA mutations and other gene mutations contribute to ARHL; a significant load of acquired mtDNA mutations have been shown in ARHL auditory tissue. Recently, several lines of evidence indicate that oxidative phosphorylation dysfunction is the common pathway of ARHL. 17 Amounts of correlative data from morphological, bioenergetic, biochemical, and genetic studies that support the mitochondrial theory of aging 18 are as follows. Normal metabolism causes ROS production by the electron transport chain; subsequently, ROS, which are generated inside the mitochondria, damage lipids, proteins, 19 and the nucleic acids in mitochondria. 20 As a result, the ROS-induced mtDNA mutations lead to the synthesis of functionally impaired respiratory chain subunits, which cause respiratory chain dysfunction and augmented ROS production. 21 Finally, the accumulation of this damage ultimately leads to permanent age-related mitochondrial dysfunction, which in turn leads to apoptosis and contributes to ARHL. 22 Thus, we may speculate that inherited gene mutations or the ROS-predisposed mtDNA variants play a major role in the progression of ARHL.
Previous investigations have reported that caloric restriction (CR), which retards several aspects of the aging process in multiple species and slows the progression of ARHL in C57BL/6J mice, reduces the levels of apoptosis in the cochlea. 23 Apoptosis may play a key role in the age-related decline of physiological function in the aging cochlea through suppressing the age-related increases in capase-3, capase-9, 24 and Bcl-2 family members, such as Bak.
In the present review, we begin by summarizing the initiating events by which ROS induce apoptosis. Then we outline several types of mtDNA mutations and other gene mutations (Table 1) and review recent insights into their mechanism (Fig. 1), and emerging themes in their pathogenesis followed by discussions of the current preventative strategies. Finally, we discuss the challenges and opportunities that lie ahead for ARHL.
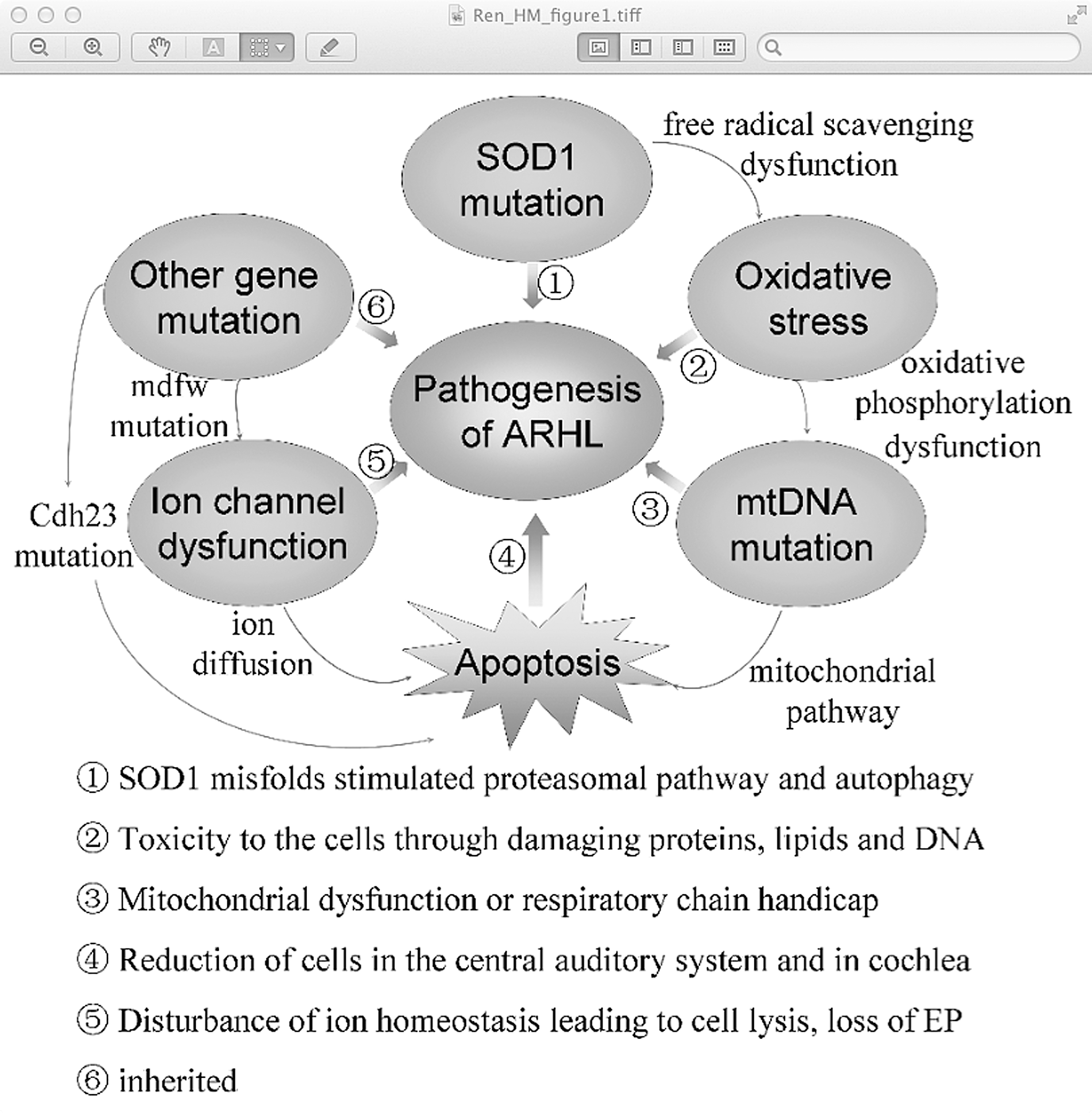
The mechanism of age-related hearing loss (ARHL). The SOD1 mutation, as an initial event accelerating ARHL through stimulating the proteasomal pathway and autophagy, directly leads to free radical scavenging dysfunction, which in turn stimulates mitochondrial (mt) DNA mutation. mtDNA mutations are not only toxic to the auditory cells by damaging proteins, lipids, and DNA in cells but also results in cell apoptosis through the mitochondrial pathway, which untimately leads to ARHL. Other gene mutations such as the Cdh23 mutation may be harmful to the stereociliary tip link and cause hair cell apoptosis from the base to apex, leading to early onset of ARHL. Meanwhile, the mdfw mutation leads to the plasma membrane Ca2+ ATPase2 dysfunction, finally resulting in cell apoptosis and even the appearance of ARHL.
SOD1, superoxide dismutase 1; mtDNA, mitochondrial DNA; ARHL, age-related hearing loss; NIHL, noise-induced hearing loss; mt-Tr, a single nucleotide insertion in the tRNA-Arg gene; mdfw, modifier of deaf waddler ① nucleotide positions 13,447–13,459 and 8470–28,482 ② deletion junction (bp 8103, bp 12,937–12,952); ③ tRNALeu(UUR) ④, an adenine at position 9826 in mt-Tr.
ARHL: Oxidative Phosphorylation Dysfunction-Related Apoptosis
Since mitochondrial disease was first described by Rolf Luft and his colleagues in 1959, 25 more than 150 distinct genetic mitochondrial syndromes have been defined, such as lactic acidosis, skeletal myopathy, blindness, sub-acute neurodegeneration, intestinal dysmotility, and peripheral neuropathy, especially deafness. 26 Mitochondria are a major source of ROS-induced oxidative damage, which is consistent with the mitochondrial free radical theory of aging in which aging results from the accumulated oxidative damage caused by ROS, originating from the mitochondrial respiratory chain. 27 Various of studies have shown that mitochondrial ROS play a critical role in cochlear aging, even in ARHL. 22
As our ability to define the genetic and environmental bases of mitochondrial disorders has accelerated, new questions have arisen. How can ROS result in mitochondrial dysfunction? The main pathway relevant to the two events awaits further study. Apoptosis may make a great contribution to the age-related decline of physiological function in the cochlea. 28 Moreover, the progression of ARHL in C57BL/6J mice increases the levels of apoptosis in the cochlea and reduces the levels of the mitochondrial pro-apoptotic Bcl-2 family member Bak. 29 It has also became clear that either Bcl-2 and other members of the family are essential for apoptosis. 28,30,31 Bak and Bax, either directly or indirectly, induce the release of cytochrome c and mitochondrial fragmentation. 32 Subsequently, cytochrome c diffuses from the inter-membrane space into the cytosol 33 and leads to caspase activation 34,35 and apoptosis. All of the factors mentioned above that are sufficient to induce or inhibit the apoptosis pathway are similar to what has been described for Alzheimer disease, amyotrophic lateral sclerosis (ALS), and so on. 36,37 Aggregates of mtDNA mutations that promote apoptosis may be a central mechanism that drives ARHL during mammalian aging. By interfering with various cellular functions, such as intracellular transport 38 and mitochondrial metabolism, cellular stress ultimately gives rise to mtDNA mutations.
SOD1 mutation : Initial event accelerating mtDNA mutation
Superoxide dismutase 1 (SOD1), composed of 153 amino acids, is a cytosolic enzyme that converts the superoxide radical to hydrogen peroxide and O2. SOD is the only eukaryotic enzyme that is capable of detoxifying ROS and is involved in free radical scavenging. 39 A study showed that sod-12345 Caenorhabditis elegans worms (a sod quintuple mutant that lacks all five sod genes) exhibit a specific sensitivity to ROS and use multiple compensatory mechanisms to maintain low levels of oxidative damage. 40 Previously, the mechanism of SOD1 in the degenerative diseases such as ALS has been illustrated. Mutant SOD1 misfolds and is targeted for degradation through ubiquitylation. Additionally, mutant SOD1 seems to have adverse effects against the cell's degradation machinery by impairing the proteasomal pathway and autophagy. 41,42
Various types of genetic study have shown that deletion of SOD1 leads to accelerated ARHL with underlying pathologies of severe SGN loss, reduced size of the stria vascularis, and accelerated out hair cell (OHC) loss. 43 Meanwhile, another investigation demonstrated that SOD1 appeared to be important for the survival of cochlear neurons and the stria vascularis; however, even half the amount is sufficient and an over-abundance does not provide much protection from ARHL. 39 Interestingly, genetic over-expression of SOD1 has not been shown to provide a protective benefit against ARHL. 39,44 Further interventions may focus on increasing the activity of anti-oxidant enzymes or decreasing the production of free radicals. This endeavor could be an effective method for reducing age-related degeneration of sensory cells in the cochlea. Collectively, all of these studies may lead to a conclusion that it is critical to maintain the balance of anti-oxidant/oxidative stress for ARHL.
mtDNA mutations
mtDNA mutations are hypothesized to cause aging and have been found to increase in a variety of tissues in aging individuals. 45 The “common” deletion (CD) was initially identified in patients with Kearns–Sayre syndrome 46 and was detected in heart muscle, brain, liver, and skeletal muscle of elderly individuals. 47 –49 The results from animal experiments and human autopsy studies have suggested that somatic mtDNA mutations, including CD, are considered to be the significant underlying factor in the progression of ARHL. 47,50,51 Previous studies have revealed that the mtDNA 4977-bp mutation, known as the CD, plays an important role in ARHL. 51 Numerous investigations have been conducted to demonstrate the association between elevated mtDNA mutation levels and ARHL. It is reported that 14 out of 17 patients with presbycusis had detectable levels of the mtDNA 4977-bp deletion in archival temporal bone, whereas only eight of the 17 human specimens presented the mtDNA 4977-bp deletion with normal audiograms. 52 However, the mtDNA 4977-bp deletion also occurs in young people and in children with hearing loss. All of those reports indicated that the CD occurred not only in elderly individuals with normal hearing but also in young people. So, we surmise that the effect of the CD on ARHL is still poorly understand. Meanwhile, a similar mtDNA 4834-bp deletion that resulted in the down-regulation of the activity of anti-oxidant enzymes and the up-regulation of lipid peroxidation was also detected to occur in naturally aging rats. 53 However, several studies have indicated that the mtDNA 4834-bp and mtDNA A3243G deletions 54 increase in aged rats and have a great relevance with ARHL, 55 whereas Kong reported that the mtDNA 4834-bp deletion cannot directly induce the hearing loss, but can act as a predisposing factor that can greatly enhance the sensitivity of the inner ear to the aminoglycoside antibiotic. 56 Subsequently, the association between the mtDNA A3243G deletion, diabetes mellitus, and hearing loss was confirmed in population studies of patients with diabetes. On the basis of these controversial results, we proposed that the mtDNA mutation might also occur in people without ARHL and the CD might not directly induce ARHL. Therefore, the exact mechanism of mtDNA deletion on ARHL needs further basic and clinical studies.
Gene mutations
Cadherin 23 (CDH23), which is encoded by CdH23, is an important constituent of the hair cell tip link in the organ of Corti. Thus, mutations in CdH23 lead to mechano-electrical transduction dysfunction and are associated with ARHL in humans and C57BL/6J mice. 57 –59 A series of loci on chromosome 10, such as mdfw, Ahl, and waltzer (v), are also associated with ARHL. The mdfw locus on chromosome 10 modifies the hearing loss phenotype in mice in heterozygous for Atp2b2 ( plasma membrane Ca2+ ATPase2 gene). 60 The genetic complementation tests indicate that mdfw may be allelic with Ahl. 61 The Cdh23 gene is localized in the mdfw and ahl interstitial genomic region and responsible for ARHL. 57 The predisposition to early-onset ARHL that is conferred by Cdh23 depends on the effects of mitochondrial mutation mt-Tr in A/J mice. 17 Combination of the “accelerating alleles” with Cdh23 is sufficient to induce ARHL onset. A study of Cdh23nmf308/nmf308 in mice reported that the CdH23 mutation may be harmful to the stereociliary tip link and causes hair cell apoptosis from the base to apex, which leads to early onset of ARHL. 59 All of those reported data indicated that homozygosity with respect to CdH23 significantly increases the susceptility to ARHL, but it is not the only cause for its pathogenesis and symptoms in phenotypic manifestation. We may conclude that the occurrence of ARHL not only attributed to the Cdh23 mutation but also to the additive or stochastic interactions of secondary factors with Cdh23. The genetic architecture of ARHL may provide a paradigm for predisposition to ARHL in humans and defines a presbycusis model to explore therapeutic avenues, such as stem cell therapy.
In addition to the discovery of the mtDNA mutation, predisposing genetic factors are also responsible for most forms of ARHL. Ahl is the first and major gene (locus on chromosome 10) that affects late-onset of ARHL and has been mapped in mice. 62 The Ahl gene has an epistatic interaction with A/J-derived mtDNA, wherein homozygosity for the predisposing Ahl allele is a prerequisite for the manifestation of the mtDNA effect on hearing loss. 17 Furthermore, it may increase the susceptibility of mice to noise-induced hearing loss (NIHL) so that preventative measures can be taken, such as minimizing exposure to loud sounds. Therefore, Ahl not only directly affects the onset of ARHL but it also indirectly plays a role in NIHL by interacting with mtDNA and up-regulating the susceptibility to loud sounds. The same author later reported the genetic mapping of Ahl2 (a locus on chromosome 5) that contributes to the difference in ARHL onset times between NOD/LtJ (NOD) and C57BL/6J (B6) mice. Moreover, the influence of Ahl2 on ARHL seems to be more strain specific than that of Ahl, and may be restricted to NOD-derived strains. 63
The molecular identification of ARHL-contributing genes, such as Ahl and Ahl2, in experimentally amenable inbred mouse strains will help to identify similar genes in human populations. In 2007, a study demonstrated that Ahl3 (a locus on chromosome 17) exists within the region between D17Mit274 and D17Mit183 and affects not only ARHL but also renders in B6 mice more susceptible to NIHL, which is highly similar to Ahl. The interaction of Ahl1 or Ahl3 on ARHL is attributed to the resistant alleles in either locus, such as persephin (Pspn), neurturin (Nrtn), and solute carrier family members (Slc25a23 and Slc5a7). 64,65 Studies of knockout mice for anti-oxidant enzyme copper/zinc superoxide dismutase (Cu/Zn SOD) and cellular glutathione peroxidase (GPX) genes and ion regulators showed they were more vulnerable to ARHL. Of note, the candidates of Ahl3 factors described above seem to be functionally related to Cu/Zn SOD and GPX and ion regulator genes. 66,67 Meanwhile, Ahl4, a locus on chromosome 10 contributes to the early-onset, rapidly progressing hearing loss of A/J mice because of its earlier and more severe hair cell loss in A/J mice than in B6 mice during the first 5 months of age. 68 The authors also reported the effects of Ahl5, and Ahl6, 69 Ahl8, 70 Phl1, and Phl2 71 on ARHL and indicated that they can be tested as candidate genes for ARHL susceptibility. Gene identification could provide entry to the molecular pathways of pathogenesis and pathophysiology of ARHL and may contribute to the development of diagnostics, which suggests a possible means of preventative interventions and therapies for human presbycusis.
Other factors inducing ARHL
Ca2+
An in vitro study detected that overstimulation of isolated outer hair cells (OHCs) will up-regulate the level of cytoplasmic [Ca2+]. 72 Phosphatidylinositol phosphate kinase type 1γ (PIPKIγ) is a key enzyme in the generation of phosphatidylinositol 4,5-bisphosphate (PI[4,5]P2) and is primarily responsible for the synthesis of the receptor-regulated phospholipase C (PLC)-sensitive PI(4,5)P2 pool in the cell syncytia that supports auditory hair cells. Homozygous knockout mice lacking this enzyme die post-natally within 24 hr. PIPKIγ catalyzes the phosphorylation of phosphatidylinositol 4-phosphate (PI[4]P) at the D-5 position of the inositol ring, ultimately resulting in PI(4,5)P2 formation, a minor glycerophospholipid of the inner leaflet of the plasma membrane involved in cell signaling cascades. Then, PI(4,5)P2, either combined with the PI(4,5)P2-binding proteins or acting as the substrate of PI(3,4,5)P3 kinase, 73 is converted into diacylglycerol and inositol 1,4,5-triphosphate (IP3) generated by PLC-dependent hydrolysis of PI(4,5)P2. IP3 activates Ca2+ efflux from the endoplasmic reticulum, raising the cytosolic free Ca2+ concentration ([Ca2+]i) by binding to its receptors (IP3R) 74 or through gap junction (GJ) channels such as P2x7 receptors (P2x7Rs) and pannexin-1 channels, 75,76 which have been implicated in adenosine triphosphate (ATP) secretion from glial cells during Ca2+ wave propagation 77 and amplification of astrocytic ICS. 78 The co-expression of Px1 and P2x7R produces a signaling complex that, when activated by ATP, in turn opens a cation channel, which creates a trans-membrane pathway that is sufficiently large to allow the passage of large-molecular-weight fluorescent dyes (up to 900 Da), and cell lysis. 79,80
The formation of vast functional syncytia coupling transfer of ions, signaling molecules, and nutrients through GJ channels contributes to the functional maturation of the cochlea. 81 Therefore, alterations of IP3R Ca2+ signaling in non-sensory cells of the developing cochlea are linked to impaired hearing acquisition in mice. The PIPKIγ deficiency in mice results in a sizeable and spatially graded down-regulation of IP3R Ca2+ signaling along the sensory epithelium of the cochlea. On the basis of the primary role played by PIPKIγ in IP3 signaling, we surmised that a reduction of PI(4,5)P2 levels may affect the development of hearing capability. Accordingly, transgenic mice with PIPKIγ deficiency study showed that PIPKIγ plays a key role in the inner ear and that it has an essential function for the acquisition of hearing. 82
The disturbance of neuronal calcium homeostasis has long been suggested to be an important factor underlying age-related impairment of neuronal function. 83 Numerous investigations have focused on the effect of T-type calcium channels in ARHL of SGNs. B6 mice homozygous or heterozygous for a null allele of the α1H calcium channel subunit can significantly slow age-related auditory brainstem response (ABR) threshold shifts, after a dramatic decrease in age-related SGNs loss. 38 Thus, anti-epileptic drugs that block T-type calcium channel may lead to better hearing thresholds in females during aging, 84 which suggests that calcium dysregulation contributes to ARHL.
The large conductance voltage- and Ca2+-activated potassium (BK) channel, which is composed of the pore-forming α-subunit co-expressed with the auxiliary β1-subunit, has been suggested to play an important role in the signal transduction process of cochlear IHC. Consistent with the phenotype elicited by the pharmacologic blockade of KCNQ4 channels, BK channel α-subunit (BKα−/−) and β1-subunit (BKβ1−/−) knockout mice were accompanied by OHC dysfunction. The human BK-coding slo1 gene mutation as a susceptibility factor for progressive deafness is similar to KCNQ4 potassium channel mutations. 85,86
K+
Several K+ channels and transporters occur in various compartments of the stria vasularis with a K+-rich extracellular fluid known as endolymph, which is essential for preserving the sensory structures and supporting transduction and has been shown to be involved in EP formation. Maintaining the endolymph homeostasis is critical to sustain auditory functions. Na+, K+-ATPase, and Na+, K+, 2Cl− co-transporter (NKCC) are localized on the basolateral membranes of marginal cells 87 –90 and are linked to the maintenance of the inner ear osmotic balance 91 and ARHL. 92 The loss of EPs, which results from a decrease of Na, K-ATPase activity, is thought to be the causative factor of ARHL in aged Mongolian gerbils. Subsequently, the loss of ATP in the strial cells may lead to permanent cell damage and/or dysfunction of the Na+-K+-ATPase pump, with the resulting in a long-term reduction of the EP.
Vibration of the basilar membrane opens the mechanosensitive channels in hair-cell stereocilia; the ensuing K+ influx 93 and the permeation of Ca2+ amplify the motility of hair bundles. 94,95 After K+ exits hair cells on their basolateral surfaces, it must cycle back to the endolymph through a pathway comprising perilymph, supporting cells, the spiral ligament, and the stria vascularis, including MCs. 90,96,97 One study 98 reported that intrastrial potential (ISP) represents the K+ diffusion potential, which also contributes to the evoked potential (EP) across the apical membranes of intermediate cells through Ba2+-sensitive K+ channels. EP originates from the ISP, which indicates that the potential difference across the membranes of intercellular space (IC) can be explained largely as K+ diffusion potential and underlies the ISP 99,100 interference with any of these elements to interrupt hearing.
ATPα is the sole Na pump α-subunit in auditory organs. ATPα preferentially localized to the scolopale cell ablumenal membrane and consistent with a role in pumping K+ ions into the scolopale cell en route to the receptor lymph, which resembles its contribution to generating vertebrate inner ear endolymph. Furthermore, the nrv2 and nrv3 of ATPα subunits show cell-type-specific expression and functions to scolopale cells and neurons, respectively. ATPα is required for hearing. 101
Fibrocytes
Spiral ligament (SL) fibrocytes in the lateral wall of the cochlea supply K+ to the strial pump vial gap junctions by recycling back into the stria vascularis. Na-K-ATPase–rich strial marginal cells could pump the K+ ions into the cells, with K+ subsequently flowing down its concentration gradient into the endolymph. The fibrocytes will take K+ from the perilymph to the endolymph. 102 A recent study in 129S6 and BALB/c mice found that dysfunction of stellate fibrocytes could disrupt a medial K+ recycling pathway that serves to remove potassium from around the inner hair cells along this pathway. Perhaps a widespread degeneration of cells in the cochlea can critically impair ion and fluid homeostasis. 13
Inflammation
Once the inner ear is exposed to bacteria, endotoxin and an abundance of monocytes and macrophages are transported into the perilymph. 102 Additionally, the increase in the generation of ROS combined with the imbalance of anti-oxidant enzymes such as manganese superoxide dismutase (MnSOD) and GPX may cause a persistent inflammation. 7,103 The release of ROS will activate the cell signaling pathways including mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) kinase (MEK), and ERK phosphorylation and activation of specific transcription factors (e.g., nuclear factor-κB [NFκB], AP-1) are also increased along with the release of ROS. In response to ROS, mitochondrial dysfunction, increased gene expression of death receptors, and/or their ligands (tumor necrosis factor-α [TNFα], FasL) appeared. All of the factors activated by the ROS in the perilymph might act directly on the vascular endothelial cells and the SL fibrocytes, which will produce various inflammatory mediators. Finally, those pro-inflammatory cytokines could cause the SL fibrocyte damage that is seen with cochlear inflammation. It is reasonable to think that chronic inflammation is one of the causal factors that might trigger premature ARHL.
Therapeutic Strategies for ARHL Derived from the Biological Pathogenesis
The development of effective therapeutic strategies to prevent or treat ARHL has proven to be a relatively difficult progress, which is demonstrated by the lack of the restorative medicines and technologies. To the best of our knowledge, ARHL is primarily attributed to the inner ear HC, subsequently leading to SGN degeneration. The ensuing HC loss is irreversible because of its limited capacity to regenerate in mammals. Therefore, we would like to explore the advantages and limitations associated with the currently available strategies for inner ear restoration, i.e., stem cell-based therapy, gene therapy, anti-oxidant defense, and operant training.
Stem cell-based therapy
Stem cell-based regenerative therapy is a potential cellular therapeutic strategy for patients with incurable brain diseases. The stem cell transplantation strategy has received significant attention as an alternative therapy for ARHL in pre-clinical studies. The cellular and biological mechanisms underlying the observed effects of a stem cell-based treatment in an animal model need to be determined. 104 First, it might be possible to replace lost neurons or cochlear cells by transplantation of stem cell–derived cells that have been pre-differentiated in vitro to various stages of maturation. Second, cell replacement might also be achieved by inducing endogenous stem cells in the adult CNS or cochlear to form new neurons, HCs, and SGNs. Finally, the grafted stem cells and their derivatives could induce optimal recovery that could also be of clinical value by releasing therapeutic molecules for immunomodulation, trophic actions, neuroprotection, and stimulation of angiogenesis. 105
In a study of senescence-accelerated mice (SAMP1), 106 allogeneic bone marrow cells derived from BALB/c(H-2K) mice were transplanted in the SAMP1 mice with ARHL. Allogeneic bone marrow transplantation (BMT) was found to prevent the development of immunological dysfunction, hearing loss, and apoptosis of the spinal ganglion cells by replacing abnormal hematopoietic stem cells (HSCS) with normal HSCS. Finally, the author demonstrated that the accelerated presbycusis resulted not from the defects in the cochlea, but from the defects in the HSCs, which are differentiated into immunocompetent cells. Although several studies have revealed that autoimmune mechanisms participate in the occurrence of ARHL, this factor may be one of the mechanisms that resulted in ARHL; however, it is not the only factor. Just as we mentioned before, ARHL is attributed to the pathology of the CNS and peripheral system including cochlea.
The conclusions in the SAMP1 mice study and the other studies seem to be contradictory. 107 We have accessed the capability of intra-cerebral embryonic neural stem cell (NSC) transplantation for C57BL/6J mice with ARHL, showing that auditory function of C57BL/6J mice can be partially restored by intra-cerebral embryonic NSCs transplantation. Just as we surmised, despite the achievements in the treatment of the degenerative diseases described above, ARHL is more suitable for stem cell therapy because of its relatively better environment for graft survival without inflammation compared to stoke and rare complications concerning of Parkinson disease after regeneration therapy. In addition to the transplantation of stem cells into the central nervous system, several studies focused on the regeneration of the cochlear cell by stem cell transplantation. It has been observed that transplantation of various types of stem cells derived from embryonic, neonatal, and adult animals have been observed to differentiate into HC or SGNs containing similar markers and proteins with functional properties. Four weeks after transplantation of induced pluripotent stem cells (iPSCs) into the cochlea of C57BL/6J mice, most parts of the cochlea exhibited the settlement of iPSCs. 108 Previously, embryonic stem cells (ESCs) have also been used to generate neurons that make synaptic contacts with HC. 109 Recently, a novel protocol demonstrated that two types of otic progenitors that differentiated from human (h) ESCs can engraft, differentiate, and significantly improve auditory-evoked response thresholds after being transplanted into an auditory neuropathy model. 110
Despite the effectiveness we currently observed, future studies must face the challenge of finding the mechanisms underlying the observed improvements: (1) How can we make best use of stem cells that may be better compliant mechanically and structurally with native tissues and integrate the transplanted tissue with the native tissue without immunological rejection? 111 (2) How can we make them grow where and how we want? (3) What is the correlation between the grafted cells and the native cells and how do the responses to downstream networks affect the computations of behavioral functions? 112 More solid pre-clinical studies have to be conducted prior to further patient applications.
Gene therapy
Gene therapeutic strategies have long been considered to be a promising and novel approach for delivering therapeutic genes or RNA interference (RNAi) to treat ARHL; gene strategies have been applied in the inner ear pathologies both in vivo and in vitro. On the basis of the reported findings, the Atoh1 gene is most commonly applied to stimulate HC production and to provide modest improvements in hearing function. 113 A number of genes driving the morphogenesis of inner HC differentiation, especially reduction and delayed deletion of Atoh1, result in the progressive degeneration of the majority of the HC within 3 weeks after birth. 114,115 Interestingly, RNAi-mediated gene suppression is typically superior to the methods of gene knockdown or direct gene therapy for its exquisite sensitivity to the target sites. 116 The transfer results of the Akv-1 and apoA-II genes were previously reported. Both of these genes are potential genetic markers, indicating that are potentially useful for ARHL diagnosis. 117,118 However, the therapeutic genes can also be mediated by stem cells and then delivered to animal models. Compared with direct gene transfer, an advantage of cell-based gene delivery is that production of the trophic factor continues even if the disease process destroys the endogenous cells. In an in vitro study, an adenovector-mediated Atoh1 gene induced HC-like cells after being delivered into human umbilical cord mesenchymal stromal cells (hUCMSCs). 119 Nevertheless, vectors used to deliver the gene in animal models have been the subject of intense speculation and controversy for several years, as they played an important role in the efficiency of gene therapy. Further investigations need to take into account their characteristics of stability and target selection, off-target effects, immune activation, and toxicity to inner ear. 120
Anti-oxidant defense
Caloric restriction
We are all aware that CR, which entails reducing food consumption by 25%–60% without malnutrition, 121 consistently extends the life span and health span of a variety of species and ARHL in mammals. 23,122 As we have discussed in this review, ARHL is linked to accumulated ROS-induced oxidative damage after mitochondrial function decline. 123 Simultaneously, the accumulation of mtDNA mutations may also initiate apoptosis in the development of ARHL. 124
Numerous studies have suggested that CR can reduce the oxidative damage, enhance anti-oxidant defense, 125 up-regulation of the sirtuin pathway, and stress response-induced up-regulation of heat shock proteins and so on. Recent studies have suggested that the loss of critical cells through apoptosis is an important mechanism of ARHL in mammals and that CR can retard apoptotic cell death in the mouse cochlea and prevents the late onset of presbycusis. 23,126
The SIRT family of genes helps regulate gene silencing, DNA repair, rDNA recombination, apoptosis, 29,127 and mitochondrial energetics. 128 Shinichi Someya reported that CR may directly stimulate Sirt3, a member of the sirtuin family, to deacetylate; this, in turn, activates mitochondrial isocitrate dehydrogenase 2 (Idh2). Idh2 is an enzyme that converts 2-oxoaldehyde dehydrogenase (NADP+) to nicotinamide adenine dinucleotide phosphate (NADPH) in the mitochondria and leads to increased NADPH levels and an increased ratio of reduced oxidized/glutathione, a major redox couple in the cell. CR fails to modify cell phenotypes in mice in cultured cells lacking Sirt3, which is recognized to be an essential player in enhancing the mitochondrial glutathione anti-oxidant defense system during CR. Therefore, Someya et al. proposed that Sirt3-dependent mitochondrial adaptations may be a central mechanism for aging retardation in mammals. 124 In addition to using CR to prevent ARHL, another intriguing possible approach is to introduce pharmaceutical interventions mimicking CR. Those interventions would induce the sirtuin pathway in multiple tissues by increasing the oxidative stress resistance and preventing the mitochondrial decay associated with aging. Resveratrol (trans-3,5,4′-trihydroxystilbene), a naturally occurring phytoalexin produced by a wide variety of plants, represents an attractive medicinal source in the treatment of ARHL. 129 Resveratrol has been reported to produce an anti-aging effect, 130 expansion of life span, 131 and up-regulation of mitochondrial biogenesis. 132 Resveratrol has recently been found to delay hearing loss in a F344/NHsd rat model. 133 Resveratrol therapy is a promising avenue to prevent the progression of ARHL.
Anti-oxidant enhancement
Vitamin C, vitamin E, and melatonin have been thought to be protective factors for ARHL. 126,134 Animal models that administered with anti-oxidants vitamin C and vitamin E exhibited efficiency in ARHL. 135 Melatonin, an indole hormone secreted primarily by the pineal gland, functions as a regulator of circadian rhythms and has been postulated to enhance the defense capacity of many anti-oxidative enzymes and thus to protect DNA, membrane lipids, and possibly cytosolic proteins against oxidative damage, thus functioning as an important neural anti-oxidant and free radical scavenger. 134,136,137 A cross-sectional study was carried out to determine the correlation between the hearing threshold and the plasma melatonin and ascorbic acid (vitamin C) levels. Low plasma levels of melatonin are significantly associated with high-frequency hearing loss (HL) among the elderly with ARHL. 137 Melatonin also plays a role in the appearance of autoimmune deafness by acting upon lymphocytes. 138
Lecithin, as a polyunsaturated phosphatidylcholine (PPC), is an important component of biological membranes. PPC played a rate-limiting role in the activation of SOD and glutathione, which, in turn, protect cell membranes from damage by ROS. Increasing evidence has shown that lecithin may preserve cochlear mitochondrial function and protect against hearing loss associated with aging. 139 Moreover, it has been found that aged gerbils that were chronically pre-treated with the spin-trapping compound N-tert-butyl-α-phenylnitrone (PBN) had a significantly decreased level of oxidized protein and increased glutamine synthetase (GS) and neutral protease activities as compared to young adult gerbils. If PBN administration was stopped after 2 weeks, the significantly decreased level of oxidized protein and increased GS and neutral protease activities in old gerbils will return to its primary levels. 140 Compared with the aforementioned compounds, the findings regarding SOD1 may provide an even more powerful therapy for ARHL. Mice with a complete loss of SOD1 exhibit an acceleration of ARHL, whereas there is no significant effect on ARHL from either the reduction or the over-expression of SOD1. 39,135 Thus, in a study in which there was chronic administration of SOD1 mimetics, the Eukarion experimental compounds EUK-189 and EUK-207 almost completely reversed the cognitive deficits and increase in oxidative stress. Particularly, protein oxidation was completely improved. 140 The demonstration of these methods signified the potential application of one or more medicine compounds to treat ARHL.
Operant training
ARHL is currently described as a degenerative disease with characteristics of age-related hearing decline associated with difficulty in speech discrimination, recognition, sound detection, and localization. Fortunately, both the animal and human studies have shown that extended training mostly improved discrimination thresholds 141 through the change in auditory pathways and the amygdala. 142,143 The improvement is specific to the trained stimuli, which suggests that plasticity occurs in areas of the brain that have relevant information about low-level (for example, physical) properties of the stimuli. 144 It has been shown that attention and reward can modulate perceptual learning and influence its specificity. 145
The perceptual and cognitive declines resulting from ARHL cannot be explained solely by a dysfunction of peripheral sensory organs, and they frequently translate to slowed perceptual processing and difficulty in accurately identifying stimuli. 146 A recent study on age-related cortical processing deficits in the primary auditory cortex of aging and young rats showed a nearly complete reversal of the majority of previously observed functional and structural cortical impairments. A significant positive impact on the frequency resolution and signal-in-noise processing of a series of cortical processing abnormalities appeared in both young and aged rats after a variety of operant training, for example, oddball training, whereas the response modulation was clearer and could also be the major contributor to the restoration of temporal selectivity in the aged auditory cortex. The authors concluded that age-related cognitive decline is a tightly regulated plastic process and demonstrated that most of these age-related changes are, by their fundamental nature, reversible. 147 The underlying mechanisms with operant training may be specific refinement of cortical representations, 148 changes in protein expression, 149 and alterations in the cortical inhibitory circuitry 150 and supporting glia. 151
Uncertainty surrounding other therapeutics
Various investigations have applied salicylate to treat noise-induced hearing loss. Although one author has mentioned that the possibility of actually improving hearing in the inner ear with ARHL is an exciting idea with significant clinical implications, but there is insufficient evidence to support the hypothesis to date.
Challenges and Opportunities
With the rapid development of technology, ARHL turns out to be a sophisticated disorder and this may explain why there are insufficient therapeutic strategies for ARHL until now. Moreover, the mutations in SOD1 have paved the way for mtDNA mutations, which lead to the apoptosis in the auditory system. The discovery of Cdh23, Ahl, and other gene mutations may also give rise to progressive hearing loss in several patients as well as the entire families. Additionally, not only the gene mutations but also the diversity of ion diffusion through ion channels all contribute to the pathogenesis of ARHL. Therefore, the exact mechanisms underlying the peripheral degeneration and central degeneration are multi-factorial, thus posing a great challenge for both pre-clinical and clinical research on ARHL. Nevertheless, ARHL also offers many opportunities for therapeutic intervention.
Over the past few years, there has been continuous progress in our understanding the mechanisms underlying ARHL and developing approaches to ameliorate the auditory function so that the quality of life can be improved among elderly people suffering from this type of hearing loss. However, many essential questions await answers. How do we take full advantage of the cell- and gene-based therapies, drug delivery, operant training, and electrophysiologic strategies to induce the hearing curve back to its normal levels and make complete behavioral recovery in animal models? How do we evaluate the safety and efficacy of the strategies discussed above? The clinical application of these therapeutic interventions and acceptance of patients with pre-clinical evidence of efficacy and safety, as required for those strategies, is unacceptable and will delay the development of clinically useful therapies to some extent. Some patients with ARHL have few or no therapeutic options and are prepared to test any new approaches, whereas a large proportion of patients with ARHL have abandoned the chance of treatment. Thus, it is hoped that scientists and clinicians can act together responsibly to translate knowledge regarding the mechanisms of ARHL and the basic research into disease-modifying treatments that will make a substantial difference for patients.
Footnotes
Acknowledgments
This research was supported by Hunan Provincial Innovation Foundation for Postgraduate (CX2013B114).
Author Disclosure Statement
All of the authors declare that no competing financial interests exist during the process of writing and publication. The subjects' written was approved by the Second Xiangya Hospital, Central South University.