
Abstract
Diabetic micro- and macroangiopathy are devastating vascular complications that could account for disabilities and high mortality rate in patients with diabetes. Indeed, diabetic nephropathy and retinopathy are the leading causes of end-stage renal failure and acquired blindness, respectively, and atherosclerotic cardiovascular diseases (CVD) accounts for about 60% of death in diabetic subjects. As a result, the average life span of diabetic patients is about 10–15 years shorter than that of non-diabetic subjects. Furthermore, tight blood glucose control might have no more than a marginal impact on CVD in general and on all-cause mortality in particular in diabetes. Therefore, therapeutic strategies that target vascular complications in diabetes need to be developed. Recently, selective inhibition of sodium–glucose co-transporter 2 (SGLT2) has been proposed as a potential therapeutic target for the treatment of patients with diabetes because of low risk of hypoglycemia and no weight gain. Because 90% of glucose filtered by the glomerulus is reabsorbed by a low-affinity/high-capacity SGLT2 expressed in the S1 and S2 segments of the proximal tubule, blockade of SGLT2 promotes urinary glucose excretion and as a result improves hyperglycemia in an insulin-independent manner. Moreover, we have shown that SGLT2-mediated glucose overload to tubular cells could elicit inflammatory and pro-apoptotic reactions in this cell, being directly involved in diabetic nephropathy. In addition, several clinical studies have also shown that SGLT2 inhibitors could reduce blood pressure, body weight, and serum uric acid levels and ameliorate cardiovascular risk in patients with diabetes. This review summarizes the pathophysiological role of SGLT2 in vascular complications in diabetes and its potential therapeutic interventions.
Introduction
D
Two large prospective clinical studies, the Diabetes Control and Complications Trial (DCCT) and the United Kingdom Prospective Diabetes Study (UKPDS), have shown that intensive blood glucose control effectively reduces microvascular complications among patients with diabetes. 13,14 However, tight blood glucose control might have no more than a marginal impact on CVD in general, and on all-cause mortality in particular in diabetes. 15 Moreover, strict control of blood glucose may increase the risk of severe hypoglycemia and/or weight gain in diabetic patients. 13,14 Therefore, development of therapeutic strategies that target vascular complications in diabetes with low risk of hypoglycemia and no weight gain is needed.
Ninety percent of glucose filtered by the glomerulus is reabsorbed by a low-affinity/high-capacity sodium–glucose co-transporter 2 (SGLT2), which is expressed mainly on the apical membrane of renal proximal tubules. 16 –18 SGLT2 is expressed in the S1 and S2 segments of the proximal tubule but not S3 segment. 17,18 Because blockade of SGLT2 promotes urinary glucose excretion and thus improves hyperglycemia in an insulin-independent manner, selective inhibition of SGLT2 has been proposed as a potential therapeutic target for the treatment of patients with diabetes. 19 Furthermore, recent clinical studies have shown that SGLT2 inhibitors could reduce blood pressure, body weight, and serum uric acid levels and ameliorate cardiovascular risk in patients with diabetes. 20 –23
This review summarizes the pathophysiological role of SGLT2 in vascular complications in diabetes and its potential therapeutic interventions. Literature searches were undertaken in Medline using the PubMed interface. Non-English language articles were excluded. The following key words were used to select the articles: (SGLT2 or phlorizin or dapagliflozin or canagliflozin or empagliflozin or luseogliflozin or ipragliflozin or tofogliflozin) and (diabetic nephropathy or diabetic neuropathy or diabetic retinopathy or vascular complications or CVD or heart or nerve or eye or retina or atherosclerosis or hypertension or uric acid or metabolic risk).
Diabetic Nephropathy
Knockdown of SGLT2
Diabetic nephropathy is characterized by functional and structural changes in the glomerulus, such as glomerular hyperfiltration, thickening of glomerular basement membrane, and an expansion of extracellular matrix in the mesangial areas, which could ultimately lead to progression of glomerular sclerosis associated with an increased urinary excretion rate of albumin and renal dysfunction. 24,25 Although characteristic histological changes of diabetic nephropathy are diffuse and nodular glomerulosclerosis, changes within the tubulointerstitium have been supposed to be more important than glomerulopathy in terms of renal dysfunction in diabetic nephropathy. 26,27
Under hyperglycemic and/or oxidative stress conditions, a non-enzymatic reaction between reducing sugars and the amino groups of proteins, lipids, and nucleic acids has progressed at an accelerated rate to form irreversibly cross-linked moieties termed advanced glycation end products (AGEs). 28 –32 Non-enzymatic glycation and cross-linking of proteins such as collagen and elastin impair their structural integrity and function, thereby contributing to the aging of macromolecules. 28 –32 Furthermore, interaction of AGEs with the cell-surface receptor for advanced glycation end products (RAGE) elicits oxidative stress generation and evokes inflammatory and fibrotic reactions in the kidney cells, causing progressive alteration in renal architecture and loss of renal function associated with tubular injury in diabetes. 28 –32
Previously, we found that knockdown of SGLT2 by small interfering RNAs (siRNAs) significantly inhibits high glucose-induced reactive oxygen species (ROS) generation and RAGE expression in human cultured proximal tubular cells. 33 Furthermore, we found that SGLT2-mediated glucose entry into the tubular cells potentiated the cells' susceptibility toward pro-apoptotic effects of AGEs via RAGE over-expression. 33 Because apoptosis of proximal tubular cells contributes to tubular atrophy and atubular glomeruli in diabetic nephropathy, 23 –25 which are most closely correlated with declining creatinine clearance, 26,27 blockade of SGLT2 may not just improve hyperglycemia by promoting urinary glucose excretion, but could also directly inhibit glucose overload to proximal tubular cells, thereby protecting against tubular apoptosis and atrophy in diabetic nephropathy.
High glucose increased senescence-associated β-galactosidase expression levels, which were suppressed by knockdown of p21 or SGLT2. 34 Tubular senescence associated with p21 over-expression was also observed in the early phase of streptozotocin-induced type 1 diabetic nephropathy, which was ameliorated by normalization of blood glucose levels with insulin. 34 So increased reabsorption of glucose via SGLT2 could contribute to proximal tubular cell senescence in diabetes. Moreover, Vallon et al. showed that although knockout of SGLT2 did not attenuate the effects of streptozotocin on markers of renal growth or inflammation, it ameliorated the diabetes-induced inhibition of autophagy in the kidney. 35 These findings further suggest the involvement of SGLT2-mediated glucose overload in tubular cells in tubulointerstitial damage of diabetic nephropathy.
Phlorizin
In streptozotocin-induced diabetic kidneys, catalase activity is decreased, whereas glutathione peroxidase activity and 3-nitrotyrosine levels are increased, all of which are ameliorated by treatment with phlorizin, a non-specific inhibitor of SGLT. 36 Phlorizin prevented proteinuria, hyperfiltration, and kidney hypertrophy, but not glomerular hypertrophy in streptozotocin-induced diabetic rats. 37 Phlorizin treatment was also shown to reduce serum levels of AGEs, decrease albuminuria, improve renal dysfunction, and ameliorate histological alterations in the kidneys, such as mesangial expansion, glomerular basement membrane thickness, and a decreased number of podocytes in obese type 2 diabetic mice, which were associated with increased anti-oxidative capacity in the kidneys. 38 Phlorizin prevented tubulointerstitial fibrosis in type 1 diabetic nephropathy partly via restoration of the fibroblast growth factor 23–klotho axis as well. 39 These observations suggest that inhibition of intestinal and tubular glucose absorption by phlorizin might exert beneficial effects on experimental diabetic nephropathy.
Empagliflozin
High glucose induces expression of Toll-like receptor-4, transcriptional activation of nuclear factor-κB (NF-κB) and activator protein 1, and collagen IV expression as well as interleukin-6 secretion in human proximal tubular cells, all of which are inhibited by empagliflozin. 40 These findings suggests that is the case with SGLT2 knockdown. Blockage of glucose entry into the proximal tubular cells by empagliflozin could reduce the inflammatory and fibrotic responses to high glucose, thus attenuating the glucose toxicity in tubular cells.
Empagliflozin treatment reduced blood glucose levels, albuminuria, and renal inflammation in obese type 2 diabetic mice, which was associated with inhibition of mesangial matrix expansion and glomerular hypertrophy. 41 Empagliflozin prevented the diabetes-induced increase in the glomerular filtration rate, attenuated increases in kidney weight and urinary albumin/creatinine ratio, and suppressed inflammation in early diabetic nephropathy in type 1 diabetic Akita mice. 42 Recently, we have found that empagliflozin significantly improves hyperglycemia and glycated hemoglobin (HbA1c) and decreases expression levels of AGEs, RAGE, 8-hydroxydeoxyguanosine, and F4/80, markers of oxidative stress and macrophages, respectively, in the diabetic kidney. 43
Empagliflozin also decreased urinary excretion levels of 8-oxo-2′-deoxyguanosine (8-OhdG) and
Hyperfiltration is an early renal hemodynamic abnormality in type 1 diabetic subjects and is associated with the increased risk for development of diabetic nephropathy. 45 In type 1 diabetic patients having hyperfiltration (glomerular filtration rate ≥135 mL/min/1.73 m2), empagliflozin treatment during both clamped euglycemia and hyperglycemia conditions significantly decreased plasma nitric oxide levels and effective renal plasma flow associated with increase in renal vascular resistance, thus ameliorating hyperfiltration. 46 The observations suggest that short-term treatment with empagliflozin might attenuate renal hyperfiltration in type 1 diabetic subjects by affecting tubular-glomerular feedback mechanisms.
Dapagliflozin
Dapagliflozin acutely reduced proximal fluid reabsorption, leading to a 70% increase in early distal chloride, which could decrease the single nephron glomerular filtration rate and ameliorate hyperfiltration in streptozotocin-induced diabetic nephropathy via tubuloglomerular feedback response. 47,48 Long-term treatment with dapagliflozin ameliorated hyperglycemia, β-cell damage, and albuminuria and suppressed glomerular mesangial expansion as well as interstitial fibrosis in db/db mice. 49 Furthermore, dapagliflozin decreased macrophage infiltration, MCP-1, and TGF-β expression and oxidative stress generation in the diabetic kidneys, and also blocked high glucose-elicited inflammatory and oxidative stress reactions in cultured tubular cells. 49
Tofogliflozin
Tofogliflozin dose-dependently suppressed glucose entry into tubular cells. 50 High glucose exposure (30 mM) for 4 and 24 hr significantly increased oxidative stress generation in tubular cells, which were suppressed by the treatment of tofogliflozin or the anti-oxidant N-acetylcysteine (NAC). 50 MCP-1 gene expression and apoptotic cell death were induced by 4 hr and 8 days of exposure to high glucose, respectively, both of which were also blocked by tofogliflozin or NAC. 50 These observations suggest that suppression of SGLT2-mediated glucose entry into tubular cells by tofogliflozin could inhibit oxidative stress generation and inflammatory and pro-apoptotic reactions in high glucose-exposed tubular cells. Tofogliflozin suppressed plasma glucose and HbA1c and preserved pancreatic β-cell mass and plasma insulin levels in db/db mice, which was accompanied with attenuation of glomerular hypertrophy and albuminuria. 51 When female mice were treated orally with several types of SGLT2 inhibitors from 1 day before transurethral inoculation with Candida albicans, the number of viable C. albicans cells in the kidneys was significantly increased by 10 mg/kg dapagliflozin or canagliflozin, but not tofogliflozin. 52 Dapagliflozin and canagliflozin significantly increased urine glucose concentration up to 24 hr after drug administration, whereas tofogliflozin it increased only up to 12 hr. 52 Therefore, risk of urinary tract infection may be lower in tofogliflozin-treated diabetic subjects than those with dapagliflozin and canagliflozin due to its shorter duration of persistent glucosuria. 52
Luseogliflozin
Control of hyperglycemia with luseogliflozin but not insulin prevented the fall in glomerular filtration rate and reduced the degree of glomerular injury, renal fibrosis, and tubular necrosis in type 2 diabetic rats, thus suggesting the pleiotropic effects of luseogliflozin in diabetic nephropathy. 53 Combination therapy of luseogliflozin and lisinopril was more protective against diabetic nephropathy than either treatment alone. 53
Others
Ursodeoxycholic acid decreased SGLT2 over-expression in type 1 diabetic nephropathy and subsequently suppressed oxidative stress in the kidneys. 54 SGLT2 expression was increased in hypertensive rats, which was inhibited with the treatment of ramipril or losartan. 55 Losartan prevented an increase in SGLT2 expression in type 1 diabetic rats as well. 56 These observations suggest that there is a pathological crosstalk between the renin–angiotensin system and SGLT2 in diabetic nephropathy.
As mentioned above, there is accumulating evidence to show that inhibition of SGLT2 might protect against high glucose-induced tubular cell damage and renal injury in experimental diabetic nephropathy. 57,58 However, recently, a spontaneous mutation in the gene encoding SGLT2 was discovered in a senescence-accelerated mouse prone 10 strain, a model of aging. 59 Because the mice exhibited cognitive impairments and cerebral atrophy associated with decreased glucose reabsorption in the kidney, long-term effects of SGLT2 inhibitors on the aging process should be carefully monitored in diabetic subjects.
Cardiovascular Disease
Empagliflozin
Arterial stiffness is associated with the prevalence of CVD and could predict future cardiovascular events in healthy and diseased subjects. 60,61 Empagliflozin during clamped euglycemia or hyperglycemia decreased augmentation indices at the radial, carotid, and aortic positions and carotid-radial pulse wave velocity in 40 normotensive type 1 diabetes mellitus patients. 62 Treatment with empagliflozin also reduced blood glucose levels, normalized endothelial function, and suppressed aortic thickness in streptozotocin-induced diabetic rats, which was associated with reduced oxidative stress generation in aortic vessels and in blood. 63 Furthermore, as is the case in streptozotocin-induced type 1 diabetic nephropathy, 43 macrophage infiltration and expression levels of AGEs, RAGE, and pro-inflammatory markers, such as MCP-1 and ICAM-1, in the aortae of diabetic animals was prevented by empagliflozin. 63 Given the contribution of quantitative and qualitative alterations of collagens and elastin by AGEs to decreased elastic properties of the vessels and increased arterial stiffness, 60,61 empagliflozin might protect against vascular injury in diabetes partly by inhibiting the AGE–RAGE axis. Empagliflozin treatment for 10 weeks significantly decreased blood glucose and oxidative stress levels in cardiovascular tissue of db/db mice and inhibited cardiac macrophage infiltration, which was accompanied with amelioration of vascular function, cardiac interstitial fibrosis, pericoronary arterial fibrosis, and coronary arterial thickening. 64 Empagliflozin also significantly attenuated cerebral oxidative stress, increased brain-derived neurotrophic factor, and prevented the impairment of cognitive function and renal injury in db/db mice. 64
Safety concerns
Dapagliflozin reduced the amplitude of shortening in ventricular myocytes of type 1 diabetic rats and reduced its amplitude of the Ca2+ transients. 65 Therefore, alterations in the mechanism of Ca2+ transport may partly underlie the negative inotropic effects of dapagliflozin in ventricular myocytes. 65 Furthermore, a case report has raised safety concerns about the association of canagliflozin initiation with development of stroke in patients with multiple clinical risk factors. 66 Two randomized clinical trials with SGLT2 inhibitors are currently ongoing. 67,68 The EMPA-REG OUTCOME™ trial is a multicenter, randomized, double-blind, placebo-controlled trial to determine the long-term cardiovascular safety of empagliflozin in patients with type 2 diabetes and to investigate its potential cardioprotective effects, as well as impact on microvascular outcomes. 67 CANVAS is a double-blind, placebo-controlled trial designed to evaluate the effects of canagliflozin on the risk of CVD and to assess safety and tolerability in patients with inadequately controlled type 2 diabetes and increased cardiovascular risk. 68
No clinical meaningful QT interval prolongation was observed in ipragliflozin-, empagliflozin-, and dapagliflozin-treated subjects. 69 –71 When the long-term cardiovascular and microvascular outcomes of 10 mg dapagliflozin were added to the standard of care (SOC) versus SOC using simulation methodology, dapagliflozin reduced the incidence of myocardial infarction, stroke, cardiovascular death, and all-cause death of 13.8%, 9.1%, 9.6%, and 5.0%, respectively, and decreased the incidence of end-stage renal disease, foot amputation, and diabetic retinopathy of 18.7%, 13.0%, and 9.8%, respectively. 72
Cardiovascular Risk
Uric acid
Observational studies have suggested the association of uric acid levels with metabolic syndrome, chronic kidney disease, and CVD in humans. 73,74 Trials with SGLT2 inhibitors in patients with type 2 diabetes have reported consistent and clinically relevant decreases in uric acid levels. 22,75 –77 When plasma uric acid and fractional uric acid excretion levels were evaluated during clamped euglycemia in type 1 diabetic patients before and after SGLT2 inhibition, empagliflozin significantly decreased uric acid levels and increased its fractional excretion in urine. 78 Luseogliflozin has been shown to lower uric acid levels through alterations of uric acid transport activity in renal tubules via glycosuria. 79 These observations suggest that glycosuria rather than hyperglycemia increases uricosuria and, as a result, decreases uric acid levels in patients with diabetes.
Hypertension
Systematic review and/or meta-analysis of randomized trials showed that SGLT2 inhibitors such as dapagliflozin, empagliflozin, cangliflozin, and ipragliflozin significantly reduced weight loss and acted as osmotic diuretics, resulting in a lowering of systolic and diastolic blood pressure in type 2 diabetes. 21,22, 80,81 Although SGLTs had no significant effect on the incidence of orthostatic hypotension, osmotic diuresis-related and/or glycosuria-related adverse effects, such as urinary tract and genital infections, should be carefully monitored.
Obesity and lipid metabolism
Several clinical trials showed that SGLT2 inhibitors inhibited glucose reabsorption by the kidney, leading to increased urinary glucose excretion-mediated diuresis and caloric loss, thereby reducing body weight and improving dyslipidemia such as hypertriglyceridemia in patients with type 2 diabetes. 21,22,82 –87
Diabetic neuropathy
Luseogliflozin treatment significantly improved insulin resistance and hyperglycemia in the Goto-Kakizaki rat, a spontaneous, non-obese model of type 2 diabetes, that was associated with amelioration of peripheral diabetic neuropathy. 88
SGLT2 Expression in Diabetes
There is some controversy about the expression levels of SGLT2 in the diabetic kidney of type 1 diabetic animals; SGLT2 levels were decreased, unchanged, or increased in streptozotocin-induced diabetic rats. 35,43,54,89 However, in contrast to type 1 diabetic animals, increased SGLT2 expression was consistently observed in the kidneys of type 2 diabetic animals 56,90,91 and in tubular cells harvested from the urine of type 2 diabetic subjects, 92 the latter of which was correlated with glucose reabsorption capacity in these patients. Recently, we have found that although high glucose or AGEs do not affect SGLT2 expression in cultured proximal tubular cells, insulin increases tubular SGLT2 levels in a dose-dependent manner. 93 Insulin resistance and the resultant hyperinsulinemia may contribute to SGLT2 over-expression in the kidney of type 2 diabetes.
Conclusions
SGLT2 inhibitors promote urinary glucose excretion and as a resultant reduce blood glucose and HbA1c levels in an insulin-independent manner. 19,20 As mentioned above, improvement of cardiovascular risks, such as reduction in blood pressure, body weight, serum uric acid, and triglycerides, is also observed in type 2 diabetic patients treated with SGLT2 inhibitors. Moreover, accumulating data have suggested that blockade of glucose absorption in tubular cells by SGLT2 inhibitors might directly exert renoprotective effects on diabetes nephropathy by preventing tubular cell apoptosis and senescence, tubulointerstitial injury and fibrosis, and glomerular hyperfiltration and sclerosis (Fig. 1). Amelioration of cardiometabolic risks by SGLT2 inhibitors may lead to improvement of aortic stiffness and endothelial function as well as suppression of vascular inflammation in diabetes. Further longitudinal study is needed to clarify whether SGLT2 inhibitor-based therapies could efficiently prevent the development and progression of vascular complications, especially diabetic nephropathy and CVD in patients with diabetes. Moreover, the oxidation process has been related to the development of aging. 94 SGLT2-mediated reabsorption of glucose has been suggested to contribute to proximal tubular cell senescence in diabetes. 34 Thus, it would be also interesting to examine whether SGLT2 inhibitors could reverse the aging process in the kidney.
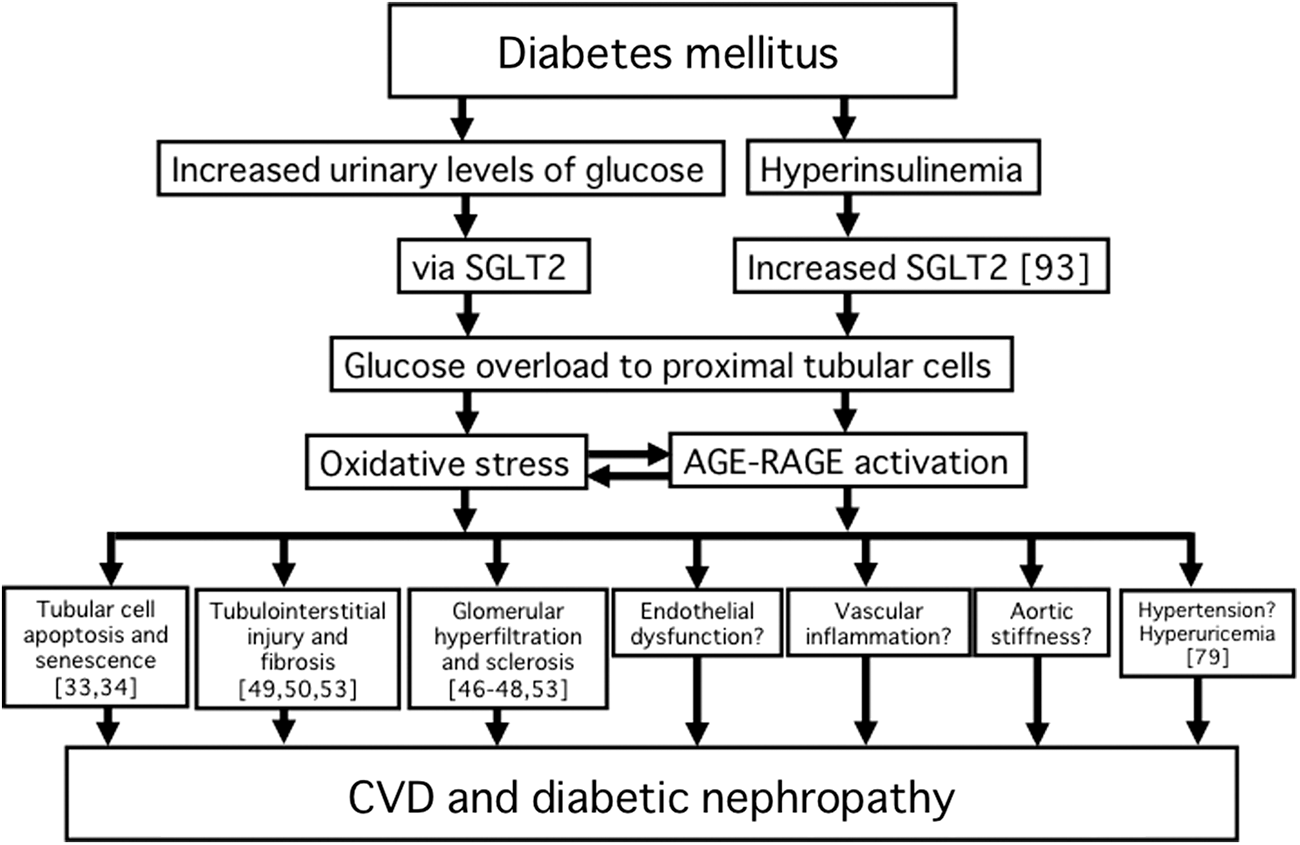
Pathological role of SGLT2 in vascular complications in diabetes. Numbers in brackets indicate reference numbers. For more mechanistic information, refer to the references. SGLT2, sodium–glucose co-transporter 2; AGE–RAGE, advanced glycation end product–receptor for advanced glycation end product; CVD, cardiovascular disease.
Footnotes
Acknowledgments
This study was supported in part by Grants-in-Aid for Scientific Research (B) (grant no. 25293127) from the Ministry of Education, Culture, Sports, Science and Technology, Japan (to S.Y.).
Author Disclosure Statement
No competing financial interests exist.
Sho-ichi Yamagishi mainly contributed to the present paper, conceptualized and designed the paper, drafted the manuscript, and took responsibility for the integrity of the data and the accuracy of the data analysis. Takanori Matsui performed a critical review for intellectual content. All authors approved the final manuscript.