
Abstract
The role of autophagy varies with the type of acute brain injury. In general, autophagy mediates a clear neuroprotective effect in intoxication caused by various psychoactive agents, subarachnoid hemorrhage and spinal cord injury. In contrast, autophagic cell death has also been reported to actively contribute to neuronal loss in neonatal hypoxic ischemic encephalopathy. However, it still remains to be determined whether autophagy pays a cytoprotective or a cytotoxic role in stroke. Previous studies focused primarily on the role of neurons rather than the role of astrocytes in brain injury. Thus, it is unknown whether modulating the autophagy flux of astrocytes contributes to improving neuronal survival after stroke. In the current study, we investigated the time course of autophagy flux in vitro using cocultured astrocytes and neurons exposed to oxygen-glucose deprivation/reoxygenation, which mimicked the process of ischemia/reperfusion. Autophagy flux of astrocytes was regulated by treatment with the autophagy inducer rapamycin, autophagy inhibitor 3-methyladenine, and the transduction of small interfering RNA against autophagy-related gene 5. In addition, AAV-GFAP-ATG7 was used to induce astrocyte autophagy flux in mice subjected to focal cerebral ischemia. We found that induction of autophagy flux of astrocytes in vitro enhanced the viability of neurons and decreased neuronal apoptosis. Furthermore, induction of astrocyte autophagy flux in mice improved neurological outcomes. In contrast, inhibition of autophagy flux in astrocytes decreased the viability of neurons and increased neuronal apoptosis. These results suggest that upregulation of autophagy flux in astrocytes may contribute to endogenous neuroprotective and neurorecovery mechanisms after stroke.
Introduction
A
Constitutive autophagy flux in neurons plays essential roles in key neuronal processes under physiological conditions at baseline levels. However, it is activated rapidly in neurons subjected to hypoxic conditions, 8 excitotoxic stimuli, 9 closed head injuries, 10 or focal cerebral ischemia. 11 A recent study has suggested that autophagy flux involved in a neuronal death pathway following ischemic brain injury. 12 However, previous studies have yielded conflict results on the role of autophagy in brain injury in that both suppression and induction of autophagy flux by pharmaceuticals reduced the severity of brain injury in rodents. 13 –15 The novel contributions of the article involve improvements in our understanding of the role of astrocyte autophagy flux in oxygen-glucose deprivation (OGD) or ischemia/reperfusion. It is the first article that addresses this question.
Astrocytes play a key role in evolution of the complexity of nervous system function. 16,17 Homeostasis of astrocytes is critical to neurons, especially during cerebral ischemia, 18 and autophagy flux plays an important role in maintaining astrocyte homeostasis. 2 It was reported that activation of autophagy after ischemia/reperfusion could be induced in neurons and astrocytes, but not in the microglial cells. 19 The dynamic balance of autophagy flux in astrocytes is necessary to ensure their basic functions. Therefore, we are very interested in investigating how different levels of autophagy flux in astrocytes affect neurons subjected to OGD and transient focal cerebral ischemia.
The importance of astrocyte–neuron interactions is supported by considerable evidence. 20,21 This study aimed to increase the neuronal survival rate following OGD and ischemic injury by regulating autophagy flux in astrocytes through administration of the autophagy inducer rapamycin (RAPA), autophagy inhibitor 3-methyladenine (3-MA), small interfering RNA (siRNA) against autophagy-related gene 5 (siATG5), AAV-GFAP-ATG7. We found that upregulation of autophagy flux in astrocytes with RAPA or AAV-GFAP-ATG7 resulted in cytoprotection and neuroprotection.
Materials and Methods
Isolation and culture of astrocytes
Primary astrocytes for cell culture were isolated from the cerebral cortex of C57BL6 mice on postnatal day 1–3 (P1–3). Briefly, P1–3 pups were sterilized in 75% alcohol and euthanized by rapid decapitation. The meninges were discarded, and the cerebral cortices and hippocampi were retained. Tissues from six mice were minced and transferred to a solution containing 0.25% trypsin (C0201; Beyotime, Jiangsu, China). Cells were extracted using single 10-minute incubations with trypsin supplemented with DNase 1 (Beyotime). Next, cells were pelleted using low-speed centrifugation (1000 rpm, 5 minutes) and resuspended in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum and a mixture of 100 U/mL penicillin and 100 μg/mL streptomycin. We plated dissociated cells on two T75 flasks evenly coated with poly-
Isolation and culture of primary neurons
Primary neurons for culture were isolated from the cerebral cortex and hippocampus of C57BL6 mice on embryonic day 18–20 (E18–20). The tissues were dissected and dissociated, and neurons were plated at a density of 20,000 cells/cm2 on 96-well or 24-well plates coated with poly-
Neuron–astrocyte cocultures
First, astrocytes were isolated from the cerebral cortex and hippocampus of newborn mice (<72 hours old) by mechanical dissociation of the tissue. The cultures were maintained in the medium described above and incubated at 37°C in a humidified atmosphere with 5% CO2. The cultures were initially seeded onto Transwell permeable supports (24-well plate; Corning, NY) and treated with 3-MA, or RAPA for 24 hours or transduced with siRNA for 72 hours. At this time, neurons were isolated from E18-20 mice, cultured as described above, and plated (5 × 104/mL) on 96-well or 24-well plates. After three rounds of washing with culture medium, the Transwell support with treated astrocytes were placed over the neurons and the cultures were maintained with neurobasal medium described above. After establishing the cocultures, the OGD/R experiments were performed. All animal procedures were performed in accordance with the Fourth Military Medical University Ethics Committee for Animal Experimentation.
Oxygen and glucose deprivation and reoxygenation
Primary neurons, astrocytes, or astrocyte–neuron cocultures were washed three times in DMEM without glucose, aspartate, glutamine, or B27 and then incubated in the same DMEM medium. Next, the cells were placed in a modular incubator chamber that was flushed with 2 L/min of a 95% N2/5% CO2 gas mixture for 15 minutes at room temperature to remove oxygen. The chamber was then sealed and placed in a 37°C incubator for 0, 15, 30, or 60 minutes. Following OGD, the cells were placed in normal medium for 24 hours of reoxygenation under normoxic conditions. For analysis of the results of the culture experiments, we used four or five cultures and each experiment was performed four to six times.
Administration of drugs
Primary astrocytes plated on Transwell permeable supports (24 well plate) were treated with 200 nM RAPA (37094; Sigma-Aldrich) or 10 mM 3-MA (M9281; Sigma-Aldrich) for 24 hours. Dimethyl sulfoxide (DMSO) at a volume identical to that of the drug treatment was used as the vehicle control. RAPA was dissolved in DMSO as a 100 mM stock solution. The 3-MA was dissolved in sterile deionized water as a 100 mM stock solution. Further dilutions were made using the culture medium.
RNA interference
siRNAs against ATG5 were purchased from GeneChem (Shanghai, China) and transduced into astrocytes to inhibit ATG5 expression. Astrocyte cultures were incubated with ATG5-specific or scramble siRNA according to the manufacturer's instructions. Briefly, astrocytes were infected with lentivirus at a multiplicity of infection of 50. The cells were used for subsequent experiments 72 hours after initiation of transfection. Immunoblotting was performed as previously described to confirm a decrease in ATG5 expression.
Staining for immunofluorescence
Cultured primary cells or brain slices were fixed with 4% paraformaldehyde at room temperature. These brains were perfused with paraformaldehyde before mice were euthanized. Primary antibodies were diluted with 5% bovine serum albumin in phosphate-buffered saline (PBS) to prevent nonspecific binding. Cells or slices were incubated with primary antibodies at least 24 hours at 4°C. The next day, after being washed with PBS, cells or slices were incubated in secondary antibodies for 1.5 hours at 37°C. Subsequently, cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, D8417; Sigma-Aldrich) for 5 minutes at room temperature. Primary antibodies and their dilutions were as follows: anti-LC3 1:500 (PM036; MBL), anti-GFAP 1:500 (3670; Cell Signaling Technology), anti-flag 1:500 (14793; Cell Signaling Technology), and anti-NeuN 1:500 (GTX30773; GeneTex). Primary antibodies were visualized with a FITC-conjugated secondary antibody against rabbit diluted 1:200 (120791; Jackson ImmunoResearch Laboratories) or an Alexa Fluor 594-conjugated secondary antibody against mouse diluted 1:500 (117783; Jackson ImmunoResearch Laboratories).
Western blots
Expression levels of LC3 and p62 in primary neurons and astrocytes were measured using western blot analysis. Briefly, soluble lysates of primary neuronal cells were mixed with sample buffer and boiled for 5 minutes at 95°C. Extracted proteins from cells were separated by 12% SDS-PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) and then electrophoretically transferred to polyvinylidene difluoride membranes. Subsequently, the membranes were blocked in 5% nonfat milk diluted in Tris-buffered saline containing 0.1% Tween 20 (TBST) for 1 hour at room temperature. Western blots were probed with the following antibodies and dilutions: rabbit anti-LC3 1:1000 (PM036; MBL), anti-p62 1:1000 (P0067; Sigma-Aldrich), anti-ATG7 1:1000 (D12B11; Cell Signaling Technology), anti-β-actin 1:1000 (CW0096M; Cwbio), or anti-GAPDH 1:1000 (CW0101; Cwbio) overnight at 4°C. After washing with TBST three times, the membranes were incubated with goat anti-mouse IgG 1:10,000 (CW0102; Cwbio) or goat anti-rabbit IgG 1:10,000 (CW0103; Cwbio) for 1 hour. Protein bands were visualized using the ChemiDoc MP Imaging System.
Lactate dehydrogenase release assay
After OGD/R, cytotoxicity was analyzed by measuring the activity of lactate dehydrogenase (LDH) released into the medium from damaged cells in the coculture system. Briefly, the medium containing detached cells was collected and centrifuged. The supernatant was utilized to assess LDH activity, which was measured using an assay kit (A020-2; Nanjing Jiancheng Bioengineering Institute, China) according to the manufacturer's instructions. LDH release was calculated as follows: LDH release (%) = (A positive − A positive blank control)/(A normal − A negative blank) × 100%. Cultures under OGD conditions for 0 minutes represented basal LDH release and were considered 100%.
Cell viability assay
Neurons, and astrocytes were cultured in 96-well plates and neuron–astrocyte cocultures were placed in a 24-well Transwell plate at 5 × 104 cells per well. OGD was performed as described above. Cell viability was measured by fluorescence chemistry using the 7Sea-Cell Counting Kit (C008-3; Shanghai 7Sea Pharmatech Co., Ltd., China) and a spectrophotometer that determined optical density at 450 nm.
Terminal deoxynucleotidyl transferase-mediated dUTP-digoxigenin nick end labeling fluorescence
Terminal deoxynucleotidyl transferase-mediated dUTP-digoxigenin nick end labeling (TUNEL) assays were performed using an in situ cell death detection kit, TMR Red (2156792; Roche, Swiss) according to the manufacturer's instructions. Briefly, after washing three times with PBS, the slides were incubated in permeabilization solution for 2 minutes on ice. They were then incubated with reaction mixture for 60 minutes at 37°C. Signals were detected using a fluorescence microscope (BX-51; Olympus, Japan).
Ischemic stroke model
All animal protocols were approved by the Institutional Animal Care and Use Committee of Fourth Military Medical University. A transient ischemia model of 60-minute middle cerebral artery occlusion (MCAO) with intraluminal filaments was performed in male C57/BL6 mice (∼24 g) using a modified version of a previously described procedure. Briefly, anesthesia was induced with 2% isoflurane in 100% O2 and maintained with 1% isoflurane. A 4-0 monofilament nylon suture with a rounded tip was introduced into the external carotid artery lumen and gently advanced into the internal carotid artery until it blocked the bifurcating origin of the MCA. The suture was removed 1 hour after MCAO. Test subjects with negligible or moderate ischemic symptoms in any behavior tests on day 1 were excluded, including lack of neurologic deficit and failure to extend the left forepaw fully.
Penumbra injection
Mice that were selected at random were anesthetized with 2% isoflurane by penumbra injection and placed in a stereotaxic frame. A glass electrode was placed stereotaxically into the right penumbra tissue (−1.0 mm posterior to bregma; 1.0 mm lateral, and 0.8 mm below the endocranium). Next, 0.3 μL of 1.5 × 1012 vg/mL AAV-GFAP-GFP or AAV-GFAP-ATG7 (Hanbio Biotechnology Co., Ltd., Shanghai, China) was injected using a 10-μL Hamilton syringe (80630; Hamilton Co., Reno, NV). After the injection, the needle was left in place for an additional 5 minutes before withdrawal. To confirm infection with AAV-GFAP-GFP or AAV-GFAP-ATG7, the brains were harvested and processed for immunostaining.
Evaluation of neurological score and measurement of infarct volume
At 24 hours after reperfusion, a 5-point scoring system reported by Longa et al. 23 was used to evaluate neurological deficits in mice: the minimum neurobehavioral score of 0 indicated that the mouse was dead, and the maximum score of 5 indicated that the mouse was normal. The assessment of neurological deficit score was performed by an observer blinded to group information. In an in vivo experiment, 54 mice were injected with virus to establish the MCAO model. Finally, 50 mice were considered to satisfy the standard. The mice were then decapitated and the brains were cut into 1-mm-thick coronal sections. The sections were immersed in 2% 2,3,5-triphenyltetrazolium chloride (TTC, T8877; Sigma-Aldrich) at room temperature for 15 minutes. Unstained areas were defined as infarcts and were measured using image analysis software (ImageJ). The percentage of infarct volume was calculated using the following formula: (total contralateral hemispheric volume − total ipsilateral hemispheric stained volume)/total contralateral hemispheric volume × 100.
Statistical analysis
All data, except for neurological scores are presented as mean ± standard error of the mean. The statistical significance of differences between the two groups was determined by an unpaired Student's t-test. For all other results containing three or more groups, an ANOVA with Tukey's post hoc analysis was used to determine significance, using GraphPad Prism 6.0 statistics software. Neurological scores were expressed as the median (range) and were analyzed with the Kruskal–Wallis test followed by the Mann–Whitney U-test for multiple comparisons.
Results
The time course of autophagy flux differs in cultured neurons and astrocytes after OGD
We set out to understand the time course of autophagy flux in cultured neurons and in astrocytes after OGD. To do that, primary neurons and astrocytes were cultured separately, and OGD/R model was established. Primary neurons and astrocytes were randomly divided into sham and OGD (1 hour)/R (0, 6, 12, 24, or 48 hours) groups. In addition, primary astrocytes were randomly divided into sham and OGD (4 hours)/R (0, 6, 12, 24, or 48 hours) groups. Western blot analysis was used to detect expression of autophagy flux marker proteins LC3 and p62. Under normal conditions, autophagy flux in neurons remained at low levels and was activated immediately upon exposure to OGD and the expression of LC3-II increased (Fig. 1A, B). After 12 hours, autophagy flux decreased to the baseline level and the expression of LC3-II increased and that of p62 decreased (Fig. 1A, C). However, autophagy flux was induced again after oxygenation for 24 hours, as well as after 48 hours (Fig. 1A–C). In contrast, OGD after 1 hour and reoxygenation did not cause the autophagy flux level of astrocytes to differ from that of the sham group (Fig. 1D–F). In this study, we observed a trend for the changes of astrocyte autophagy. Considering the robust feature of astrocytes, we next stimulated astrocyte autophagy with 4 hours of OGD. Under normal conditions, autophagy flux in astrocytes remained at high levels (Fig. 1G). It was inhibited immediately upon exposure to OGD and decreased to the lowest level after oxygenation for 12 hours (Fig. 1G–I), regarding the decreased expression of LC3-II and the increased expression of p62 (Fig. 1G–I). Autophagy flux increased after oxygenation for 24 hours as well as after 48 hours, returning to baseline levels (Fig. 1G–I). Based on these experiments, we characterized that the time course of changes in autophagy flux in neurons and astrocytes, are very different and that autophagy flux of both cell types reach a stable high level with oxygenation for 24 hours after OGD.

OGD and reoxygenation changed levels of autophagy flux in neurons and astrocytes.
Astrocytes enhance viability of neurons after OGD
To understand the role of astrocytes in neuronal viability after OGD, we tested the survival of neurons and astrocytes cultured individually, and neurons/astrocytes cocultured. Viability of neurons cultured alone was assessed at 24 hours of reoxygenation after OGD (60 minutes). Results showed that neuronal viability was reduced significantly to 65% compared with OGD for 0 minutes (100%) (Fig. 2A). We considered OGD 60 minutes/reoxygenation for 24 hours an appropriate level of stimulation. In contrast, when astrocytes that had been cultured alone were subjected to OGD (60 minutes) and after 24 hours of reoxygenation, cell viability showed no significant difference compared with those subjected to OGD for 0 minutes (Fig. 2B). This indicates that astrocytes are more resistant to OGD. Furthermore, the addition of astrocytes to neuronal cell cultures resulted in higher neuronal viability compared with that of neurons cultured alone (Fig. 2C), which suggests that astrocytes play a cytoprotective role in OGD/R.
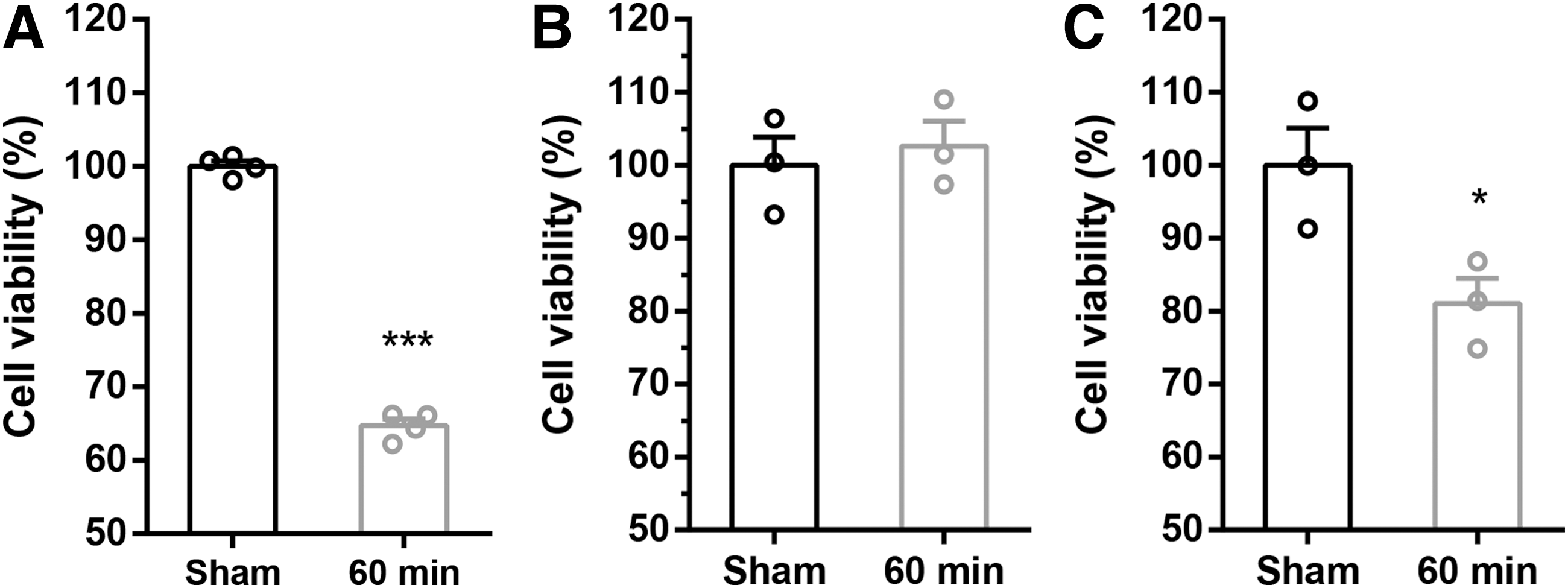
Astrocytes enhanced viability of neurons after OGD.
Induction of autophagy flux in astrocytes reduces damage to neurons subjected to OGD
To further understand the role of autophagy flux in the transition of astrocytes to neurons during OGD, astrocytes treated with siATG5, 3-MA, or RAPA as well as control cells were cocultured with neurons, and the cocultures were then subjected to OGD for 0 or 60 minutes, followed by 24 hours of reoxygenation. LDH release, and cell viability assays as well as TUNEL staining were used to evaluate effects of regulating autophagy flux in astrocytes on coculture system cytotoxicity and neuronal viability. First, we tested the ability of these treatments with siATG5, 3-MA, or RAPA to modulate astrocyte autophagy by detecting the expression of LC3-II through immunofluorescent staining and western blot analysis. Punctate LC3-positive staining was observed in astrocytes. After administration of RAPA, astrocytes positive for LC3/GFAP were activated, and such punctate accumulated in the OGD model (0, 60 minutes)/R (24 hours), as indicated by morphological examination and the number of autophagosome compared with Ctrl (Fig. 3A, C), whereas treatment with 3-MA or siAtg5 resulted in a significant reduction in autophagosome and LC3-II levels in the OGD/R model (Fig. 3A, C). To further confirm that the number of autophagosomes was indicative of enhanced autophagy flux, immunoblotting showed that LC3-II levels in the primary astrocytes (Fig. 3B, D) to be consistent with immunofluorescence. Collectively, 3-MA and siAtg5 resulted in a reduction of autophagy flux level and RAPA resulted in an increase in autophagy flux level in primary astrocytes in OGD/R model.
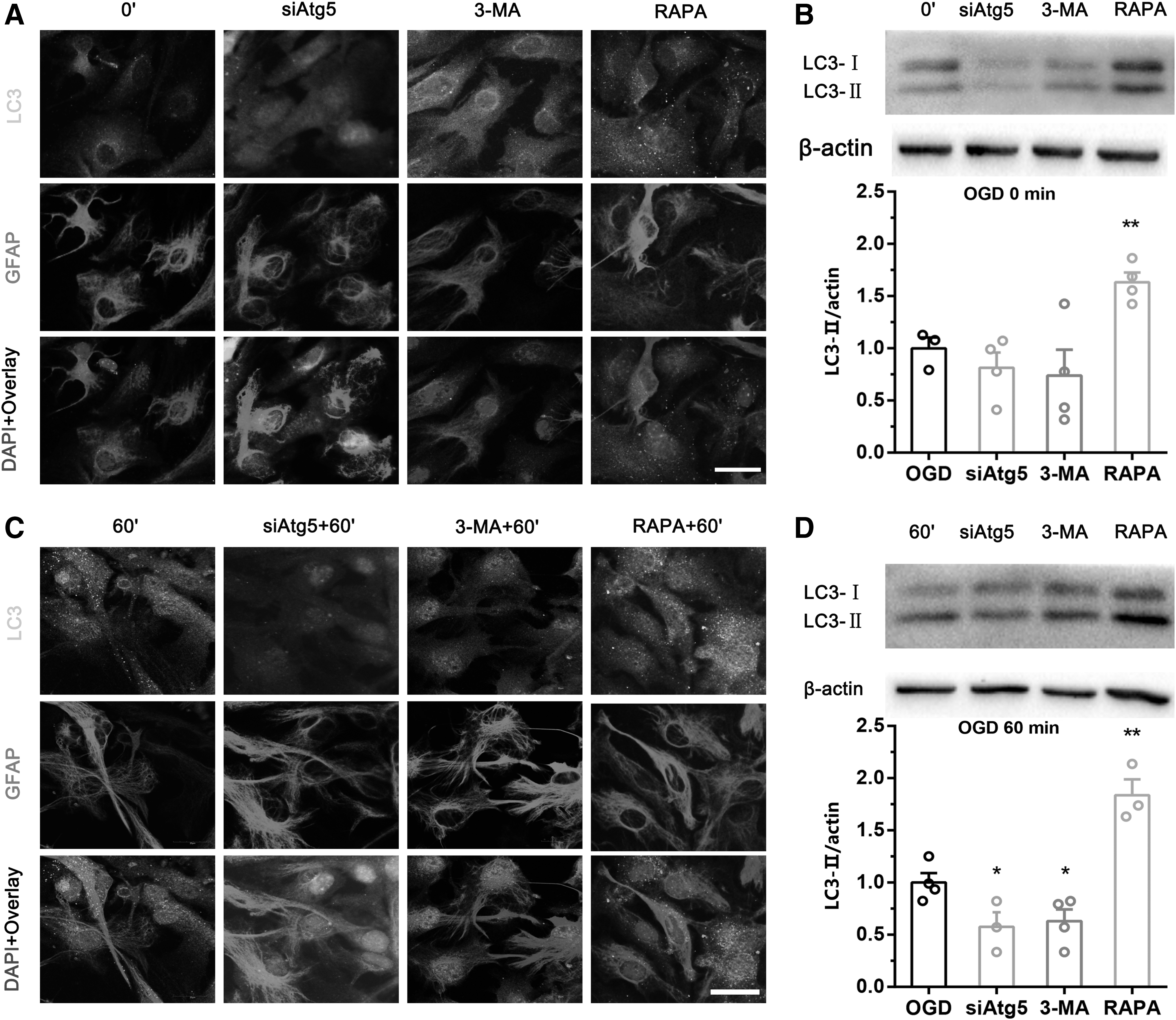
The effectiveness of regulating autophagy flux of astrocytes was observed dynamically.
No significant differences were observed between groups under normal conditions (Fig. 4A, B, E, and F). However, upregulation of autophagy flux in astrocytes with RAPA decreased apoptosis (Fig. 4C, D) and LDH levels (Fig. 4E) and enhanced viability (Fig. 4F) of neurons in coculture. In contrast, downregulation of autophagy flux in astrocytes with 3-MA or siATG5 increased apoptosis (Fig. 4C, D) and LDH levels (Fig. 4E) and decreased viability (Fig. 4F) of neurons in coculture following OGD for 60 minutes and reoxygenation for 24 hours. These results suggest that upregulation of autophagy flux in astrocytes reduces coculture cytotoxicity and promotes neuronal viability, whereas downregulation of autophagy flux in astrocytes increases coculture cytotoxicity and decreases neuronal viability after OGD.
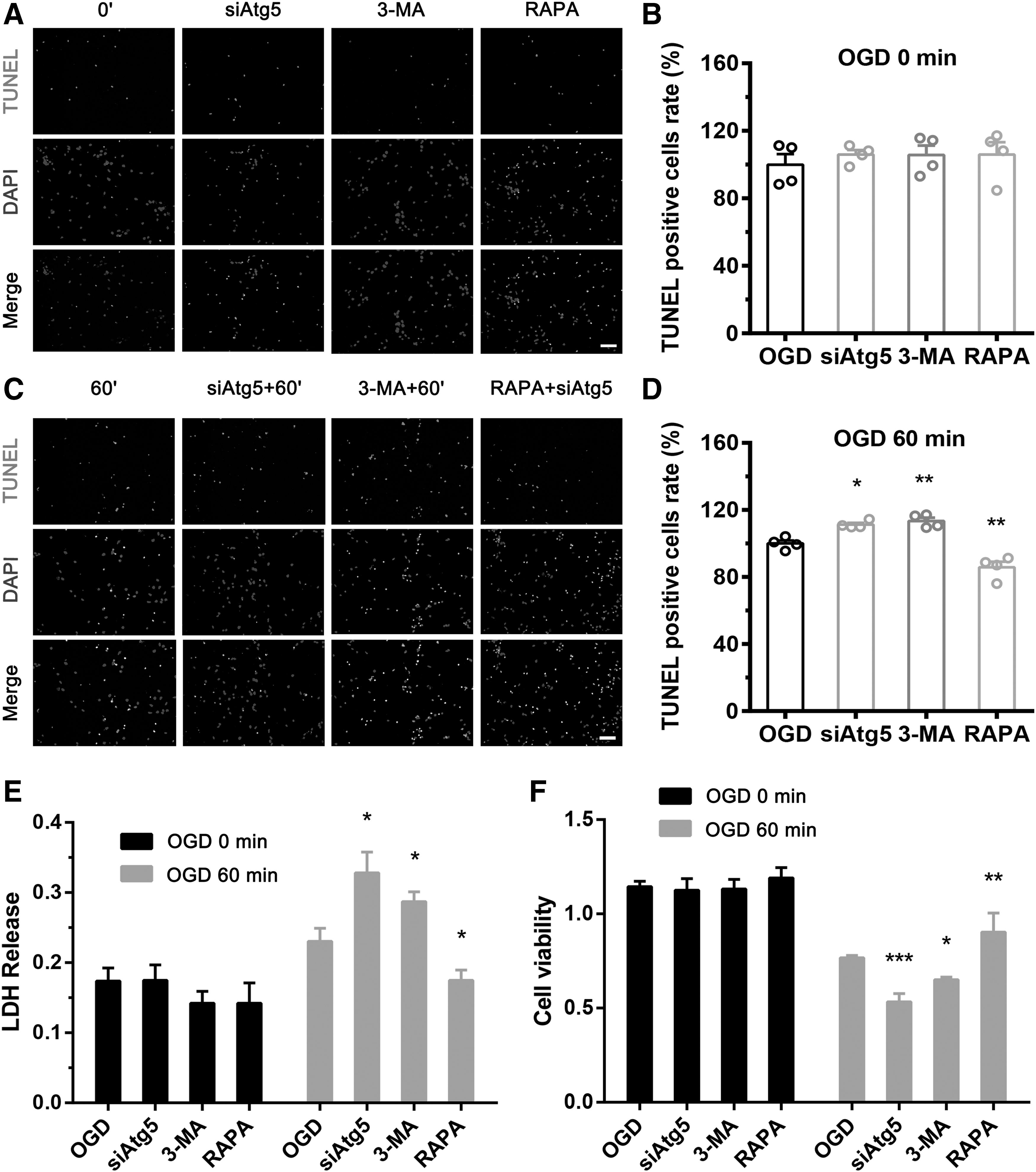
Induction of autophagy flux in astrocytes alleviated neuronal damage after OGD. From left to right, four groups of astrocytes, which were maintained under normal conditions and treated with siATG5, 3-MA, or RAPA, were cocultured with neurons. After OGD, neuronal apoptosis, cytotoxicity of the coculture system, and neuronal viability were measured by TUNEL staining, LDH assay, and CCK8 assay, respectively. The percentage of positive cells was calculated as TUNEL-positive cells (number of positive cells/total number of cells × 100%). Four groups of astrocytes had no effect on neuronal apoptosis rate
Induction of autophagy flux in astrocytes decreases infarct volume and increases neuronal survival after MCAO
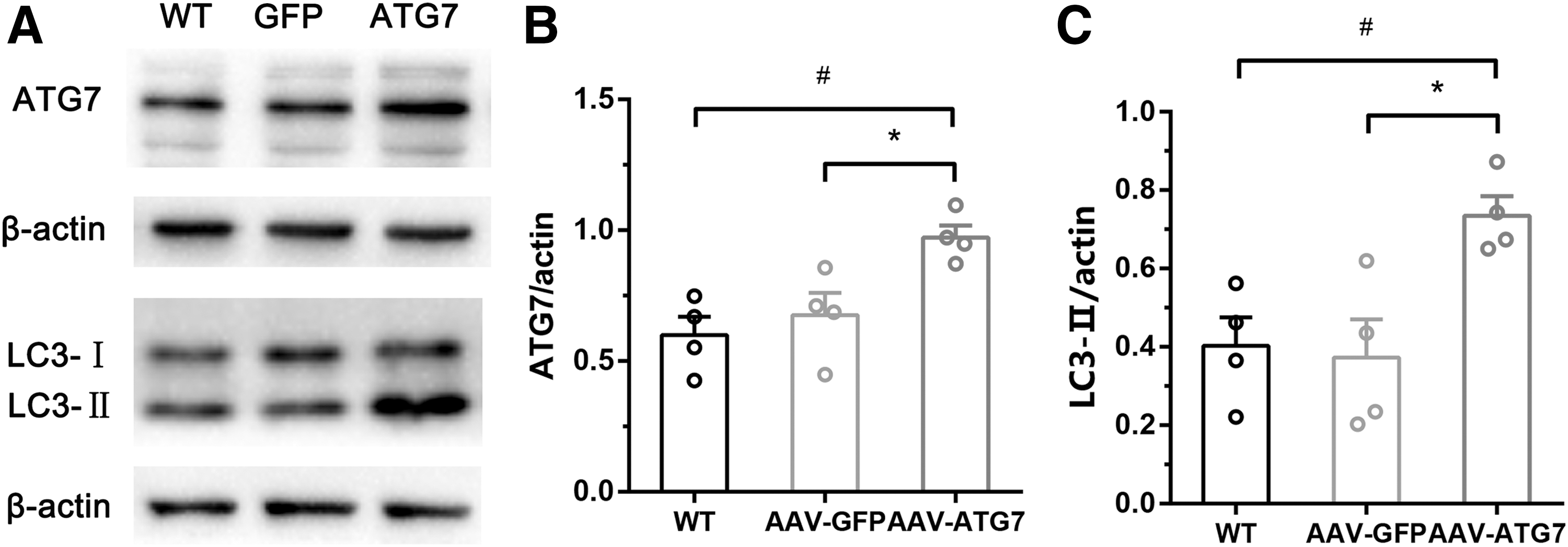
To study the effects of modulating autophagy flux of astrocytes on brain infarct volume and neuron survival in animals subjected to MCAO for 1 hour and reperfusion for 24 hours, wild-type C57BL6 mice were divided into AAV-GFAP-GFP and AAV-GFAP-ATG7 groups. An adeno-associated virus (AAV-GFAP-ATG7) that upregulates autophagy flux of astrocytes or a control virus (AAV-GFAP-GFP) was injected into the right penumbra tissue of each mouse. Three weeks later, the ischemia/reperfusion model was established. Before the model was established, we randomly chose four mice from each group and assessed the effectiveness of AAV-GFAP-ATG7 by western blot analysis. AAV-GFAP-ATG7 displayed induction of ATG7 protein, and upregulation of autophagy, as shown by the increased expression of LC3-II (Fig. 5A–C). TTC staining was used to measure the brain infarct volume of each mouse and immunofluorescence was used to assess the survival of neurons in penumbra tissues. Immunostaining confirmed the correct injection site and AAV injection of astrocytes (Fig. 6A, B). Upregulation of autophagy flux in astrocytes with AAV-GFAP-ATG7 increased the rate of survival of neurons in penumbra tissue subjected to MCAO/reperfusion injury (Fig. 6B, C) and decreased brain infarct volume compared with AAV-GFAP-GFP (Fig. 6D, F). However, upregulation of autophagy flux in astrocytes with AAV-GFAP-ATG7 did not affect neurological scores (Fig. 6E).

Induction of autophagy flux of astrocytes exerted neuroprotection after MCAO (1 hour)/reperfusion (24 hours).
Discussion
In this study, we found that autophagy flux increased in neuron and astrocyte cultures in the OGD model at different time courses, and upregulation of autophagy flux in astrocytes protected neurons subjected to OGD. Similarly, upregulation of autophagy flux in astrocytes by AAV2/9-GFAP-ATG7 played a neuroprotective role in ischemic stroke. One recent study 24 reported that neuronal Atg7 deficiency did not influence stress-related proteins in mice. Atg5 and Atg7 are involved in the induction of autophagy and autophagosome formation, without which no autophagy flux can proceed. In in vitro experiments, we used siATG5 treatment to modulate the autophagy flux in astrocytes, and we chose AAV-GFAP-ATG7 to modulate autophagy flux in astrocytes in in vivo experiments.
Brains of adult mice subjected to ischemia show changes in various biomarkers of autophagy, including inhibition of MTORC1, LC3 lipidation, degradation of p62, and upregulation of BECN and LAMP2. 25 –27 A previous study showed that the induction of autophagy with RAPA decreased infarct size in mouse models of temporary MCAO (tMCAO) and permanent MCAO (pMCAO). However, similar results were obtained with chloroquine, which is an autophagy inhibitor. 14 Moreover, 3-MA increased the loss of neurons in mice subjected to tMCAO, but it exerted a neuroprotective effect in mice with pMCAO. 28 These observations are inconsistent with the hypothesis that autophagy always exhibits neuroprotective activity in adults' cerebral ischemia. Thus, it is important to find another approach to investigate the role of autophagy flux in brain injury. In this study, we examined how compensated astrocyte autophagy flux levels affected neurons through astrocyte–neuron crosstalk and showed that upregulation of astrocyte autophagy flux levels increased the survival of cocultured neurons after OGD.
Autophagy, which is sometimes referred to type 2 cell death, is a basic cellular process that causes degradation of long-lived proteins and recycling of cellular constituents to ensure survival during starvation or other types of stress. Autophagy upregulated in many tissues during development, and is important for survival during neonatal starvation. 29 Inhibition of autophagy results in the accumulation of cytoplasmic abnormal protein inclusion bodies that can cause cell damage and neurodegeneration. Thus, autophagy plays a vital role in cell survival. 30,31 ATG5 and ATG7, which are members of the ATG family, play critical roles in autophagy, and are essential proteins in the generation of autophagy flux. 32 –34 Knockdown of ATG5 with siRNA can be used to inhibit activation of autophagy flux.
Previous research has shown that injury due to ischemia/reperfusion can significantly increase autophagy flux levels in astrocytes significantly. 35 Astrocytes outnumber neurons in the brain, 18 and they play essential roles in multiple functions of the central nervous system, including glutamate reuptake, maintenance of ion and water homeostasis, protection against oxidative stress injury, glycogen energy storage, as well as synapse formation and remodeling. 36,37 In addition, astrocytes modulate neuronal activity by releasing gliotransmitters and scavenging glutamate, are involved in synaptic support and formation, and physically contact and connect large numbers of neurons. 20,38,39 Furthermore, our results indicate that astrocytes retain more autophagy machinery function than neurons, as judged by the number of autophagosomes in the cytoplasm, as part of maintaining normal functions. More interestingly, astrocytes are migrating cells, 40 and they bridge structures, such as neurons and vasculature that otherwise cannot communicate. 41 This raises the question of whether autophagy flux in astrocytes is the key to maintaining neuronal survival after OGD or ischemic injury.
Our results suggest that neuronal rescue during recovery from OGD or ischemia is related to the level of autophagy flux in astrocytes. Immunofluorescence staining showed decreased numbers of apoptotic cells and increased neuronal viability and survival, which suggests that upregulation of autophagy flux levels in astrocytes increases neuronal survival. However, the causal relationships among these observations are unclear, and further investigations are needed to elucidate the underlying mechanism. Autophagy may serve as a homeostatic mechanism to inhibit apoptosis and limit adverse effects of chronic ischemia. 42 Induction of autophagy flux in cells decreases the activation of proapoptotic Bax, whereas suppression of autophagy through knockdown of ATG5 or 3-MA increases cellular damage, suggesting that autophagy serves as a protective mechanism against ischemia/reperfusion. 43 The critical role of astrocytes in the central nervous system is well known, 44 and effects of abnormal astrocytes on neurons have been studied widely. The stability of astrocytes is of great significance to neurons, especially in cerebral ischemia.
Previous studies focused on inducing or suppressing autophagy flux in neurons, and the results were controversial. In contrast, this study examined how regulating autophagy flux in astrocytes affects neurons. We showed that induction of autophagy flux in astrocytes reduced neuronal cell death, whereas inhibition of autophagy flux in astrocytes increased neuronal cell death. Because both 3-MA and RAPA have limited specificity for regulating autophagy flux, it is important to use more specific methods to inhibit autophagy. Thus, we also used genetic knockdown of ATG5 to downregulate astrocyte autophagy flux and AAV-GFAP-ATG7 to upregulate astrocyte autophagy flux, which are critical ATG genes.
In conclusion, this study explored how regulating astrocyte autophagy flux affects neurons, and results suggest that astrocyte autophagy flux may protect neurons from OGD or ischemic injury. However, the mechanism underlying the observed significant effects of astrocyte autophagy flux levels in astrocytes on neurons remains unknown. In general, the crosstalk between astrocytes and neurons in the context of autophagy flux level regulation remains unclear. Neuron–astrocyte interactions are vital for normal functions and viability of neurons and should be elucidated further regarding neuroprotective mechanisms such as autophagy. Nonetheless, our findings suggest that upregulation of autophagy flux in astrocytes may be a promising strategy that should be developed in depth in the hope of improving prognosis of stroke patients. It is essential to explore the pathway underlying the protective effects of upregulating autophagy flux in astrocytes in OGD-induced neuronal damage.
Footnotes
Acknowledgments
This project was supported by the Funds for International Cooperation and Exchange of the National Natural Science Foundation of China (No. 81420108013), and the Changjiang Scholars and Innovative Research Team in University, Xi'an, China (No. IRT-14R08).
Author Disclosure Statement
The authors declare that there are no conflicts of interest that could be perceived as prejudicing the impartiality of the reported research.