
Abstract
Although the health benefits attributed to urolithin A, such as anticancer, anti-inflammatory, and antioxidant effects, are based on numerous, diverse studies carried out in vitro, the biological effects of urolith A are still not entirely understood. In this study, we explored the biological effects of urolithin A using senescent human skin fibroblasts (HSFs) to determine whether urolithin A has any antiaging potential. Our results showed that urolithin A significantly increased type I collagen expression and reduced matrix metalloproteinase 1 (MMP-1) expression. Urolithin A also reduced intracellular reactive oxygen species, which may be partially due to activation of the Nrf2-mediated antioxidative response. These results indicate that urolithin A is a promising antiaging agent. Meanwhile, we noticed that the 50 μM urolithin A could cause changes in cell morphology and inhibition in cell proliferation, which were due to cell cycle arrest in G2/M phase. However, SA-β-gal (senescence-associated β-galactosidase) staining and γH2AX immunofluorescence staining showed cellular senescence status of HSFs did not change. Results of DAPI (4′6-diamidino-2-phenylindole) staining (no significant change) increased BCL2 gene expression and mitochondrial membrane potential (no significant change) after urolithin A treatment showed that the cells did not undergo apoptosis. These results provided further insights into the molecular mechanism of urolithin A. In conclusion, urolithin A showed a strong potential of antiaging.
Introduction
Urolithin A is a dibenzopyran-6-one derivative produced from ellagic acid (a product of ellagitannin degradation) by animal and human gut microbiota. 1 Following consumption of ellagitannin-rich food, such as pomegranates, strawberries, raspberries, blackberries, and walnuts, 2 the absorption of ellagitannins in animals is very low. 3 In contrast, a large amount of urolithin A is detected in animal blood and urine. 4 This poor bioavailability and the extensive gut catabolism suggest that urolithin A, rather than ellagitannins or ellagic acid, may be the actual bioactive molecules. However, in vivo evidence is still limited. Studies by Larrosa et al. and Ishimoto et al. have demonstrated the anti-inflammatory effects of urolithin A in rat and mouse models, respectively. 5,6 Ryu et al. also showed that urolithin A can extend lifespan and improve fitness in Caenorhabditis elegans. 7
Although the direct bioactivity of urolithin A in vivo remains largely unexplored, in vitro studies using specific cell models have provided clues to its mechanism in vivo. Recent research, mostly based on in vitro testing, has resulted in preliminary evidence of the anti-inflammatory, anticarcinogenic, antiglycative, antioxidant, and antimicrobial effects of urolithin A. This has pointed toward their potential role in the health benefits attributed to ellagitannin-rich foods. 8 On the other hand, the antiaging effects of urolithin A have rarely been studied. Hence, in this study, we used a replicative senescence in vitro model of human skin fibroblasts (HSFs) to study the potential antiaging mechanisms of urolithin A.
Materials and Methods
Cell culture
HSFs were purchased from Kun Ming Cell Bank of Chinese Academy of Sciences (CAS). The cells were maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% newborn calf serum (NBCS), glutamine, penicillin, and streptomycin, in a 37°C humidified incubator containing 5% CO2.
Senescence-associated β-galactosidase staining and morphology
Senescence β-gal staining was performed. 9 The assay of senescence-associated β-galactosidase (SA-β-gal) activity was carried out according to the manufacturer's protocol (#9860; Cell Signaling). The fibroblast cells, grown on a six-well plate, were briefly fixed in 1 × fixative solution containing 2% formaldehyde and 2% glutaraldehyde, for 10 minutes. Subsequently, the cells were stained overnight at 37°C with the β-galactosidase staining solution, at pH 6.0. Images were acquired by Leica DMIL-LED microscope with a DFC450C camera.
RNA extraction, reverse transcription and real-time quantitative polymerase chain reaction
All the kits and reagents were purchased from Ezbioscience (China). The total RNA of the cultured cells was extracted and prepared with a cell lysis buffer. First, the strand cDNA was synthesized with the EZ-press Cell to cDNA Kit, according to the manufacturer's instructions. The primers are listed as below: Collagen forward: 5′-GGCTCCTGCTCCTCTTAGC-3′ Collagen reverse: 5′-TTTCCACACGTCTCGGTCAT-3′ Elastin forward: 5′-CAGGTGCGGTGGTTCCTC-3′ Elastin reverse: 5′-GTAGGGCAGTCCATAGCCAC-3′
MMP1 forward: 5′-CCAGGTATTGGAGGGGATGC-3′
MMP1 reverse: 5′-CACACGCTTTTGGGGTTTGT-3′
HMOX1 forward: 5′-AAGACTGCGTTCCTGCTCAAC-3′
HMOX1 reverse: 5′-AAAGCCCTACAGCAACTGTCG-3′
NQO1 forward: 5′-GAAGAGCACTGATCGTACTGGC-3′
NQO1 reverse: 5′-GGATACTGAAAGTTCGCAGGG-3′
SOD1 forward: 5′-GGTGGGCCAAAGGATGAAGAG-3′
SOD1 reverse: 5′-CCACAAGCCAAACGACTTCC-3′
GCLC forward: 5′-GGAGACCAGAGTATGGGAGTT-3′
GCLC reverse: 5′-CCGGCGTTTTCGCATGTTG-3′
BCL2A forward: 5′-CGTACAGTTCCACAAAGGCA-3′
BCL2A reverse: 5′-ATGTGTGTGGAGAGCGTCAA-3′
BCL2B forward: 5′-GATTTTATTTCGCCGGCTC-3′
BCL2B reverse: 5′-TGATGTGAGTCTGGGCTGAG-3′ Actin forward: 5′-AGCGAGCATCCCCCAAAGTT-3′ Actin reverse: 5′-GGGCACGAAGGCTCATCATT-3′
The reagents contained 4 μL cDNA, 0.4 μL ROX reference dye2, 4.6 μL ddH2O, 0.5 μL of each primer (10 μM), and 10 μL of SYBR green mix. The reagents were pipetted into a 96-well plate, gently spun down, and placed in the 7500 Fast Real-Time PCR System (ABI). Samples were incubated at 95°C for 5 minutes, followed by 40 cycles at 95°C for 15 seconds, and at 60°C for 60 seconds. Data were collected and analyzed, and normalized with actin. Each experiment was performed in triplicate.
Western blotting
Cells were lysed in the lysis buffer (Willget, China), supplemented with protease inhibitors. Protein extracts were resolved by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel. The separated proteins were electrotransferred to NC membranes. Membranes were blocked with a blocking buffer, for 30 minutes at room temperature (RT). Membranes were then washed and incubated overnight with matrix metalloproteinase-1 (MMP1; Santa Cruz, 1:500), GAPDH (willget, 1:1000), and COL1A (Santa Cruz, 1:500) antibodies at 4°C, with gentle shaking. The membranes were washed and incubated for 1 hour with anti-rabbit secondary antibody or anti-mouse secondary antibody, at RT. The chemiluminescent bands were visualized. Protein quantification was performed with ImageJ (NIH) and the values were normalized with GAPDH. Each experiment was performed in triplicate.
Cell viability assay
The effect of urolithin A on cell viability was determined using the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Tokyo, Japan), according to the manufacturer's instructions. Briefly, 4 × 10 3 cells per well were seeded in a 96-well flat-bottomed plate. After 1 day of incubation, the cells were treated with the indicated concentration of urolithin A and then cultured in the presence of 10% NBCS in DMEM for another 1, 3, and 5 days. Following this, 10 μL CCK-8 dye was added to each well, the cells were incubated at 37°C for 2 hours, and the absorbance was finally determined at 450 nm using uQuant microplate spectrophotometer (BioTek). Reduction in the viability of urolithin A-treated cells is expressed as a percentage, compared to control cells, which were considered to be 100% viable. To ensure continual exposure to the compound, cells were transferred to a medium with fresh urolithin A on alternate days. All the compounds used in this study were dissolved in a dimethyl sulfoxide (DMSO) stock solution. The control population was treated with the corresponding concentrations, with a 0.1% DMSO final concentration. Each experiment was performed in triplicate.
Assessment of intracellular production of reactive oxygen species
Intracellular reactive oxygen species (ROS) were determined using 2′,7′-dichlorodihydro-fluorescein diacetate (DCFH2-DA; Sigma–Aldrich Co., LLC.). After 20 minutes of incubation with 5 mg/mL DCFH2-DA, the cells were washed twice in phosphate-buffered saline (PBS) and then trypsinized and seeded in a 96-well flat-bottom plate (PerkinElmer). The signal of DCF (the oxidation product of DCFH2-DA) was detected at an excitation wavelength of 485 nm and an emission wavelength of 530 nm using the PerkinElmer Victor3 V 1420-040 Plate Reader (PerkinElmer). The fluorescence observed with PBS was used to subtract the sample value from each corresponding well. The DCF fluorescence imaging was performed using the inverted fluorescence microscope (Leica DMIL-LED). Each experiment was performed in triplicate.
Flow cytometry analysis for cell cycle
Cell cycle analysis was performed by flow cytometry. Briefly, cultured cells were trypsinized in single cell suspensions and fixed with 70% ethanol overnight, at −20°C. Fixed cells were washed in PBS and stained using PI/RNase staining buffer (BD Pharmingen) for 30 minutes, at 37°C. DNA content was assessed by a FACSCalibur flow cytometer (BD), equipped with the ModiFit LT v2.0 software. Each experiment was performed in triplicate.
Mitochondrial membrane potential
To assess the mitochondrial membrane potential, cells were incubated with the mitochondria-specific cation, tetramethyl rhodamine methyl ester (TMRM, 100 nM, 35 minutes, 37°C; Invitrogen). Images were acquired by a Leica inverted microscope with excitation wavelengths of 535 nm. Fluorescence intensity was quantified using ImageJ software. Ten fields of cells were analyzed per dish, and each experiment was performed in triplicate.
4′6-Diamidino-2-phenylindole staining
HSF cells were treated with different concentrations of urolithin A for 5 days. The cell culture medium was discarded and the cells were washed twice with PBS. The cells were fixed with 4% paraformaldehyde (pH 7.4) for 15 minutes and then washed twice with PBS. Nuclei were stained with 10 μg/mL of 4′6-diamidino-2-phenylindole (DAPI; Molecular Probes), for 30 minutes, 10 and then photographed using an inverted microscope (Leica DMIL-LED microscope with a DFC450C camera) with an excitation wavelength of 470 nm.
γH2AX immunofluorescence
Cells were fixed with 4% paraformaldehyde solution at RT for 15 minutes, followed by a 5-minute permeabilization step using a wash buffer (Beyotime, China). Cells were blocked in a blocking buffer (Beyotime) for 1 hour at RT and stained with anti-γH2AX—Ser139 (1:500, #2577; Cell Signaling Technology) overnight at 4°C, followed by Alexa Fluor 488-conjugated goat anti-rabbit- (1:1000; Invitrogen) for 1 hour at RT. Cell nuclei were stained with DAPI. The fluorescence imaging was visualized by inverted fluorescence microscope (Leica DMIL-LED).
Results
The population doubling time of replicative senescent HSF was ∼4 days. Therefore, we collected data on day 5 to better observe the effect of urolithin A on cells.
Replicative senescence of HSFs
β-Galactosidase expression in senescent human fibroblasts is increased and therefore, β-galactosidase staining provides direct evidence of cell replicative senescence. 9 In vitro cultured fibroblasts reached a plateau after about 55 PDL (population doublings). 11 Therefore, we subcultured fibroblasts isolated from adult foreskin to 50–60 PDL, followed by β-galactosidase staining to determine cell senescence. More than 90% of β-galactosidase-positive stained cells were found in the culture (Fig. 1A). Hence, we used this cellular model of replicative senescence for the following experiments.
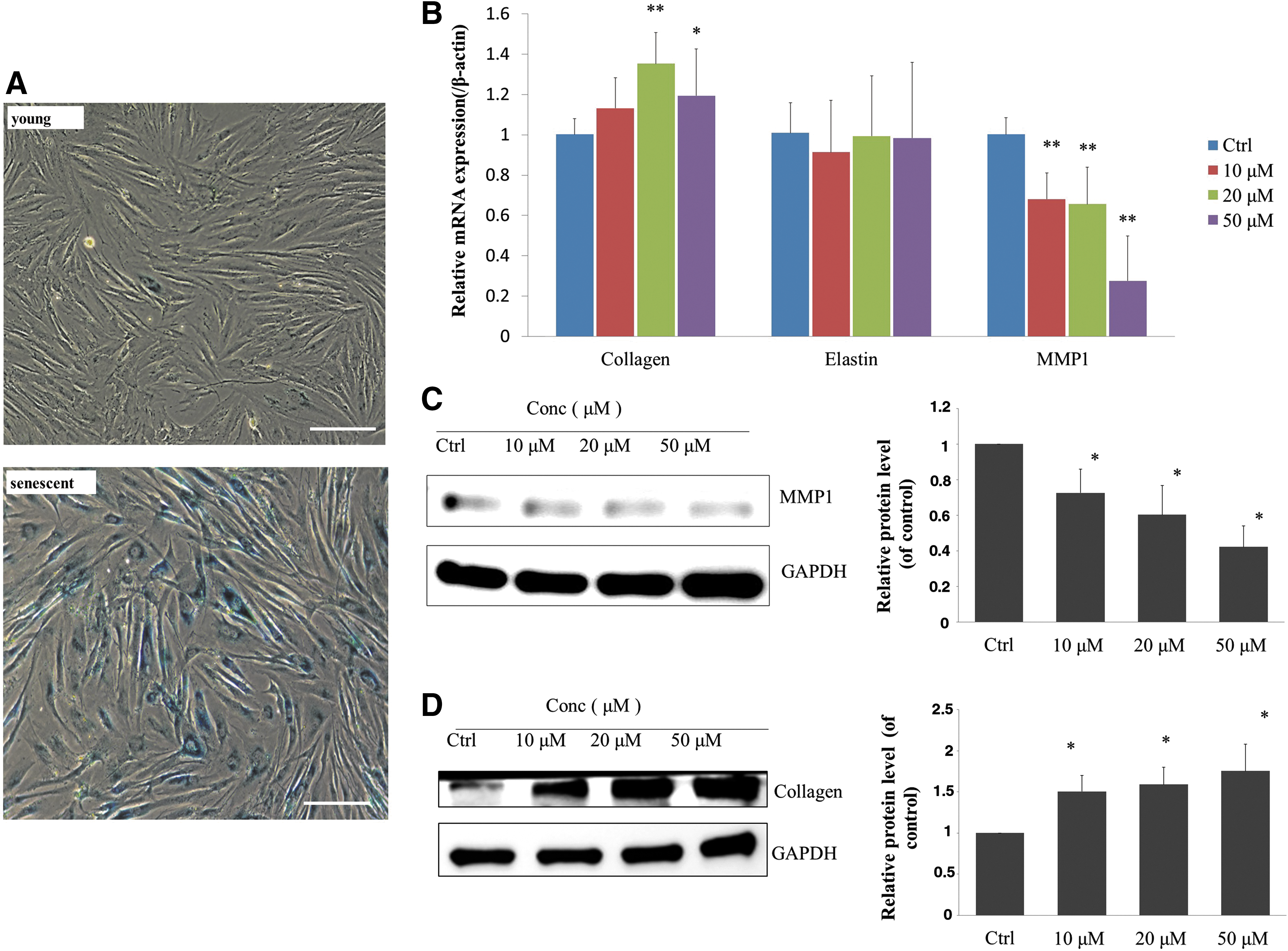
Replicative senescence of HSF and gene expression of HSF treated with urolithin A.
Urolithin A stimulated the synthesis of type I collagen expression and inhibited the expression of MMP1
Collagen is the most abundant extracellular matrix protein in dermal fibroblasts. Over 90% of the collagen in the human body is type I. 12 Elastin is an elastic protein in connective tissue that facilitates tissue restoration and helps maintain tissue morphology following stretching and contraction. 13 MMP1 is a collagenase that specifically degrades type I, II, and III collagen. 14 In naturally aging skin, type I collagen synthesis is reduced and MMP1 expression is increased. 15
We first investigated the expression of type I collagen, elastin, and MMP1 in urolithin A-treated cells. After 5 days of urolithin A treatment, no significant change in elastin mRNA expression was found. Collagen mRNA expression was significantly increased in cells treated with 20 and 50 μM urolithin A, and mRNA expression of MMP1 significantly decreased in a urolithin A dose-dependent manner (Fig. 1B). We performed Western blot analysis to compare the proteins extracted from urolithin A-treated and urolithin A-untreated cells, as well as untreated control cells. We found that type I collagen expression in urolithin A-treated cells was upregulated (Fig. 1D), while MMP1 expression was significantly downregulated (Fig. 1C). We speculated that the dose-dependent increase in type I collagen protein levels in response to varying urolithin A concentrations may be due to the downregulation of MMP1, which lowers type I collagen degradation.
Urolithin A decreased intracellular ROS generation and upregulated the expression of Nrf2 downstream antioxidant response element-response genes of HSFs
According to the ROS theory of aging, the accumulation of ROS is a major contributor. 16 Therefore, an effective method of delaying the skin aging process is to provide external antioxidants to inhibit the production of ROS or to eliminate excess ROS. 17 ROS is significantly increased in senescent cells. 18 In this study, we used DCFH2-DA to detect intracellular ROS. The replicative senescent HSFs demonstrated obvious fluorescence staining (Fig. 2B). After a 5-day treatment with urolithin A, the intracellular ROS level was significantly reduced (Fig. 2A, B), suggesting that urolithin A attenuated ROS in senescent cells.

Urolithin A reduced ROS levels and upregulated the expression of Nrf2 downstream ARE-response genes of HSFs. Intracellular ROS generation was assessed by DCFH2-DA.
We postulated that urolithin A may activate the Nrf2/antioxidant response element (ARE) pathway to stimulate quenching of intracellular ROS generation in skin fibroblasts. To test this theory, we investigated the mRNA levels of Nrf2 downstream of ARE-containing genes, such as superoxide dismutase 1 (SOD1), NAD(P)H dehydrogenase, quinone 1 (NQO1), glutamate-cysteine ligase catalytic subunit (GCLC), and heme oxygenase 1 (HMOX1), in HSF cells, after urolithin A treatment. Real-time quantitative polymerase chain reaction (qPCR) analysis revealed a significant increase in the mRNA expression of these genes in a dose-dependent manner (Fig. 2C). Overall, the results of the analyses support the idea that urolithin A regulates cellular ROS levels, at least partially through activation of the Nrf2-mediated antioxidative response.
Urolithin A did not change the fate of cell senescence
Since urolithin A increased type I collagen expression and reduced the intracellular ROS level, indicating a very strong antiaging potential, we further investigated whether cellular senescence was affected by urolithin A treatment. SA-β-gal staining of urolithin A-treated and urolithin A-untreated cells showed no significant differences (Fig. 3A). Phosphorylation of γH2AX at Ser139 is a well-established molecular marker of double-strand breaks. Inductions of γH2AX foci are commonly used as the marker of senescence. 19 We then used immunofluorescence staining to detect γH2AX foci. It showed that there was no significant difference in the signals between urolithin A-treated and urolithin A-untreated cells (data of 10 and 20 μM urolithin A-treated cells not shown).

SA-β-gal staining and γH2AX immunofluorescence staining of urolithin A-treated cells.
In addition, we noticed that 50 μM urolithin A treatment changed the morphology of cells, from long spindles to flattened cells with an increased volume, which is the typical phenotype of senescent cells. Some anticancer drugs, such as doxorubicin and diaziquone, can cause therapy-induced senescence. 20,21 We questioned if this senescence-like phenotype may indicate deep or late senescence of HSFs. Because among the senescent cells more than 90% were SA-β-gal staining positive, we explored both SA-β-gal staining and γH2AX immunofluorescence staining of young HSFs after urolithin A treatment. As shown in Figure 3B, the cell morphology also changed in young cells, but SA-β-gal staining and γH2AX immunofluorescence staining showed no significant difference in urolithin A-treated and urolithin A-untreated cells, indicating that cells were not in deep or late senescence. In addition, the morphological changes of cells were reversed within 2 days of stopping urolithin A treatment (data not shown), confirming that the effect of urolithin A on cell morphology was reversible.
All results above showed that high dose of urolithin A treatment did not have significant effects on cellular senescence status of HSFs.
High urolithin A concentration reduced cell viability and caused cell cycle arrest
We could see from Figure 3 that the cell number was reduced after a high dose of urolithin A treatment, suggesting that there may be other effects at the cellular level. To test this, we used a CCK-8 assay to determine cell viability and evaluate the effect of urolithin A at different durations (days) and concentrations of exposure. Our results showed that low doses (10 and 20 μM) of urolithin A had no significant effects on cell viability. However, urolithin A, at 50 μM, significantly reduced cell viability and the effect was exposure time dependent (Fig. 4A).
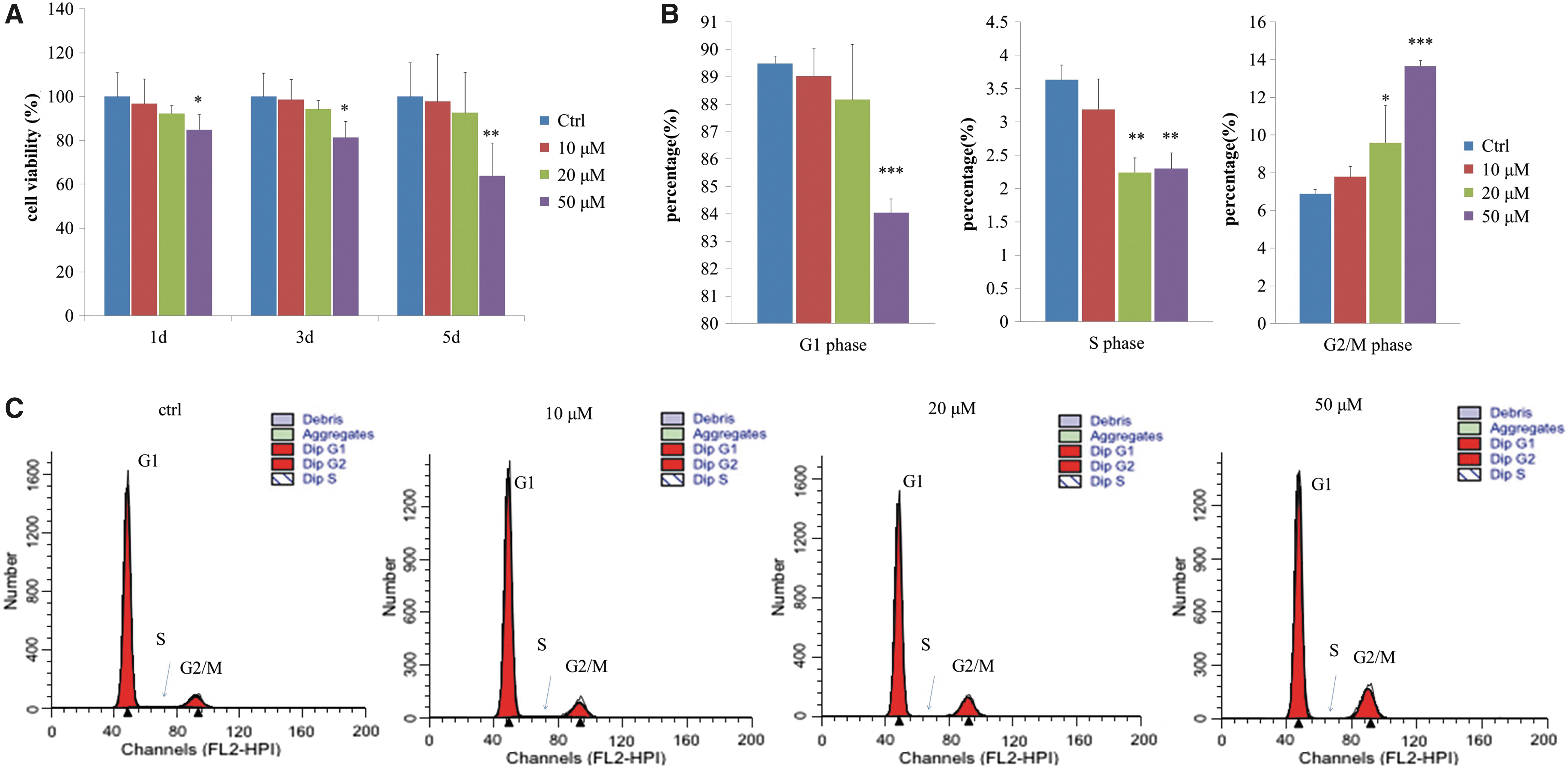
Urolithin A induces a decrease in cell viability and cell cycle arrest in a dose- and exposure time-dependent manner.
Replication of senescent skin fibroblasts slows down mainly due to cell cycle arrest in the G1 phase. 11 Cell cycle tests showed that ∼90% of the replicative senescent cells were in the G1 phase. However, while the 5-day low-dose (10 μM) urolithin A treatment had no significant effects on the cell cycle, urolithin A at 50 μM reduced the number of cells in G1 and S phases, and significantly increased the number of cells in G2/M phase (Fig. 4B, C), indicating significant G2/M cell cycle arrest. These results suggested that the inhibition of cell proliferation was due to the G2/M cell cycle arrest.
Urolithin A did not induce apoptosis of HSFs
Evidence showed that the induced G2/M cell cycle arrest may in turn lead to an apoptotic response. 22 Previous studies also showed that a high dose (50–100 μM) of urolithin A induces apoptosis in cancer cells. 23 To verify whether the cells undergo apoptosis following urolithin A treatment, we examined a few biological indicators. Apoptosis can be differentiated from necrosis by their characteristic nuclear changes. DAPI is a nuclear stain, which is observed as blue fluorescence when excited under a fluorescence microscope. As shown in Figure 5A, there were no significant changes in the nuclei after 50 μM urolithin A treatment for 5 days. BCL2 is specifically considered an important antiapoptotic protein, which is located on the outer membrane of mitochondria, where it plays an important role in promoting cellular survival. 24 Quantitative RT-PCR analysis revealed a significant increase in the mRNA expression of BCL2 in a dose-dependent manner (Fig. 5B). Reduced mitochondrial membrane potential is considered an initial and irreversible step toward apoptosis. To assess the mitochondrial membrane potential, we quantified the average mitochondrial fluorescence intensity of the cationic TMRM. 25 The results showed that the mitochondrial membrane potential of HSFs did not show any obvious change after urolithin A treatment (Fig. 5C).
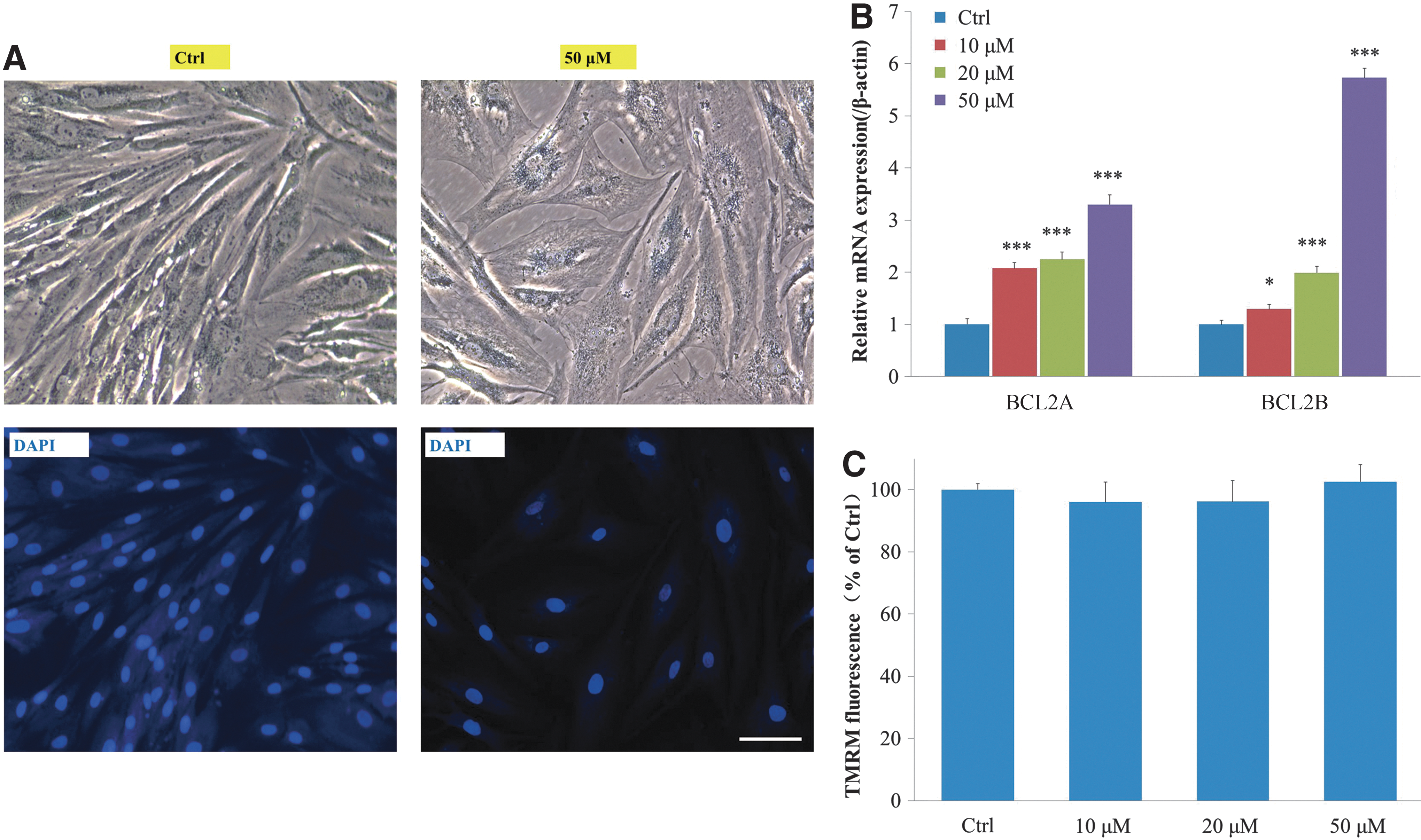
Urolithin A did not induce apoptosis of HSFs.
Together, these results demonstrated that the cells treated with urolithin A do not undergo apoptosis.
Discussion
In this study, we treated replicative senescent HSFs with urolithin A and found significant changes in the extracellular matrix gene expression and intracellular ROS. Type I collagen expression was significantly increased and MMP1 expression was significantly decreased in a dose-dependent manner (Fig. 1B, D). The urolithin A enhanced antioxidant capacity of HSFs by increasing the gene expression of phase II enzymes and antioxidant enzymes, such as SOD1, NQO1, GCLC, and HMOX1 (Fig. 2), which may be partially responsible for the elimination of intracellular ROS after urolithin A treatment. Increased type I collagen expression moisturizes the skin and reduces wrinkles. The reduction of ROS levels indicated that urolithin A had a strong antioxidant capacity. These findings suggested that urolithin A has antiaging potential.
According to the SA-β-gal and γH2AX staining results (Fig. 3), urolithin A did not change the fate of cell senescence of HSFs. However, an increased expression of extracellular matrix and a changed intracellular redox state in cell indicated that the cell quality may be improved. Although high concentration of urolithin A could induce cell cycle arrest, our results also showed cells that received urolithin A treatment did not undergo apoptosis (Fig. 5), so we speculated the cell cycle arrest and morphological changes may be due to autophagy. Ryu et al. suggested that short-term administration of urolithin A activates mitophagy in young C. elegans worms and mammalian cells, which may lead to a decrease in the mitochondrial content, while long-term urolithin A exposure results in greater mitochondrial content. This indicated that urolithin A treatment activates mitochondrial biogenesis, which improves cell quality. 7 These observations are generally consistent with our hypothesis. However we are still in need of more in-depth study of the effects of urolithin A mitophagy in skin fibroblasts.
Retinoic acid (RA) and its derivatives are effective in antiaging treatment and widely used in skin care. 26 According to the previous studies, we found that urolithin A and RA have both similarities and differences in their mechanisms of antiaging. First, both can increase type I collagen accumulation by the induction of type I collagen expression and by inhibition of MMP expression. 27,28 Second, both have antioxidant capacity. RA acting as an antioxidant may mainly depend on its own redox capacity. 29 Only a few reports showed that RA treatment can increase the activity of antioxidant enzyme. 30 Urolithin A not only acts as an antioxidant 31 but also increases the antioxidant capacity of cells by activating the expression of Nrf2 downstream ARE-containing genes, while RA has an inhibitory effect on this pathway. 32 Finally, high concentrations of RA can inhibit the proliferation of fibroblasts and increase the oxidative stress and apoptosis of human dermal fibroblasts, 33 while high concentrations of urolithin A can cause cell cycle arrest and inhibit cell proliferation without apoptosis induction. In summary, urolithin A is a promising agent for antiaging and can be an alternative of RA in cosmetics.
In vitro antioxidant activity studies of urolithin A on H2O2- or PMA (4β-phorbol-12β-myristate-R13-acetate)-induced cells showed that IC50 values of intracellular generation of ROS were ∼10–50 μM. 34 We adopted these concentrations on senescent HSFs in our research and got promising results. However, there are no reports of urolithin A conjugate occurrence in skin tissues of animals that consume foods with rich ellagitannins or ellagic acid. 8 To achieve a certain concentration on the skin surface to achieve antiaging of skin, urolithin A can be added as a raw material to antiaging cosmetic formulations that can be used daily. Taking percutaneous absorption into account, it is difficult to reach the above concentration on dermal fibroblasts, so our next topic is to explore the minimum effective concentration of urolithin A on HSFs. Our research focuses on dermal fibroblasts, on the top of dermis is epidermis; it is also necessary to explore the influences of urolithin A on keratinocytes. To assess the potential side effects of its long-term application, toxicology safety tests should be performed, such as skin irritation, skin sensitization, phototoxicity, reproductive & developmental toxicity, genotoxicity, and so on. The next step is to determine the clinical efficacy of urolithin A formulation in improving skin appearance compared with base formulation without urolithin A.
Footnotes
Acknowledgments
This work was supported by the National Key R&D Program of China [2016YFF0203204]; Technology Projects of General Administration of Quality Supervision, Inspection and Quarantine of the People's Republic of China [2016IK219]; and National Key R&D Program of China [2017YFC1200500].
Author Disclosure Statement
No competing financial interests exist.