
Abstract
Brain aging is an intricate and natural phenomenon exclusively characterized by oxidative stress, accumulation of oxidatively damaged macromolecules, and alterations in structure and function of neurons that further increase the risk factor for most of the neurodegenerative diseases. In addition, age-dependent defective autophagy has also been implicated to favor the pathogenesis and prevalence of the neurological diseases. Therefore, the development of strategies that delay aging and the concomitant neurological disorders remain elusive. Thus, the present study was undertaken to investigate the effect of rapamycin-induced activation of autophagy on aging-related oxidative stress, cell death, neuroinflammation, and neurodegeneration in rat brain. Our data demonstrated the significant age-related oxidative stress, apoptotic cell death, elevated inflammatory response, and reduced level of markers associated with rejuvenation and neural integrity, including the activities of ion channel transporters (Na+/K+-ATPase and Ca2+-ATPase) and acetylcholinesterase in the brain of old aged rats. Furthermore, rapamycin (0.5 mg/kg b.w. for 28 days) induced activation of autophagy provided significant protection to aging rat brain by reducing the aging-induced oxidative stress, apoptotic cell death, and markers of neurodegeneration. Thus, our data confirmed that autophagy plays a pivotal role in delaying brain aging plausibly by maintaining the cellular homeostasis, and structural and functional integrity of cells in the brain.
Introduction
A
Activated microglia mediated inflammation is another factor in aging and the levels of inflammatory mediators such as interleukins (IL-1β and IL-6), tumor necrosis factor-alpha (TNF-α), cytotoxic complement components, reactive oxygen species (ROS), and nitric oxide have been reported to increase with advancing of age even in the absence of acute infection or other physiological stress. 8,9 In addition, oxidative products that accumulate in microglia during aging make them vulnerable to oxidative damage and cell death that may also contribute to the neurodegeneration. 10
Autophagy is a highly conserved catabolic process that continuously degrades and recycles oxidatively damaged cell organelles and protein aggregates to maintain cellular homeostasis. 11 Therefore, defective autophagy during advancing of age has been suggested to further contribute to the accumulation of abnormal protein aggregates and, thereby, increase the risk of neurological diseases. 12 Furthermore, autophagy has also been suggested to control various biochemical and molecular events of neuronal physiology, 13 as well as aging of the cells. 14
The mammalian target of rapamycin (mTOR), a serine/threonine protein kinase, is a central player in the regulation of growth, survival, proliferation, and motility of the cells through altering protein synthesis. 15 Several studies suggest that mTOR signaling pathway is involved in the onset and progression of diabetes, cancer, and aging. 16 Moreover, mTOR also negatively regulates autophagy process, 17 and reduced mTOR signaling has been reported to extend the life span of yeast, worms, flies, and mice. 18 The inhibition of mTOR can be achieved by several drugs, including rapamycin. 19 Rapamycin, an antibiotic derived from Streptomyces hygroscopicus, is a FDA-approved immunosuppressant drug that also activates the autophagy process through selective inhibition of mTOR. 19
The pharmacological interventions to alleviate the consequences of aging on brain are limited and not yet rewarding and also associated with significant risk of adverse effects. Therefore, the development of strategies that delay aging and the concomitant neurological disorders remain elusive. The present study was undertaken to investigate the effect of rapamycin-induced activation of autophagy on aging-associated oxidative stress, cell death, neuroinflammation, and neurodegeneration in rat brain.
Materials and Methods
Materials
All the chemicals and reagents, including 2,4,6-Tri(2-pyridyl)-s-triazine (TPTZ), reduced GSH, 2,4-dinitrophenylhydrazine (DNPH), dithiobis nitro benzoic acid (DTNB), and TRIzol reagent, were Sigma-Aldrich Chemical Co. (St. Louis, MO) unless otherwise stated. cDNA Synthesis Kit and Polymerase Chain Reaction Kit were procured from New England Biolabs. All the antibodies were procured from Abcam. Rapamycin was purchased from TCI Chemicals (India) Pvt. Ltd. All other chemicals were of analytical grade available from Merck and SRL. Autoclaved deionized water was used in all the experiments.
Animals and treatment
Male Wistar rats of age 4 and 18 months weighing 140 ± 20 g and 300 ± 30 g, respectively, were used in the study. Rats were bred and maintained in the animal house of our department. Hygienic conditions were strictly maintained to rear the animals in controlled conditions (25°C ± 2°C and relative humidity 55% ± 15%) with a 12-h light/12-h dark cycle. The animals were fed with a normal laboratory diet of nutrient rich pellets containing total energy as fat, protein, and carbohydrates (Pranav Agro Industries Ltd., Pune, India) and had free access to drinking water. All experiments were carried out in sync with the guidelines laid down by the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Ministry of Environment and Forests (Government of India), New Delhi, India, and experimental procedures were in accordance with the Animal Care and Ethics Committee of University of Allahabad, Allahabad, India.
Animals were randomly allocated to the following four treatment groups having 12 rats in each group.
Young control: Young rats were given single oral administration of vehicle solution (0.9% NaCl solution containing 0.5% DMSO) for 4 weeks daily.
Young rapamycin: Young rats were given single oral administration of rapamycin (0.5 mg/kg b.w.) for 4 weeks daily.
Old control: Old aged rats were given single oral administration of vehicle solution (0.9% NaCl solution containing 0.5% DMSO) for 4 weeks daily.
Old rapamycin: Old aged rats were given single oral administration of rapamycin (0.5 mg/kg b.w.) for 4 weeks daily.
The dose of rapamycin (0.5 mg/kg b.w.) was selected on the basis of our earlier published reports. 20
Preparation of brain tissue homogenate
Following the completion of the treatment schedule, the rat brains were isolated after decapitation, washed in ice-cold saline, weighed, and homogenized in 10% ice-cold medium containing 50 mM Tris-HCl (pH 7.4) and 300 mM sucrose using homogenizer (Potter Elvehjem glass-teflon homogenizer) following the protocol of Garg et al. 21 The brain tissue homogenates were centrifuged at 1000 g for 10 minutes at 4°C to remove cell debris and nuclei. The resulting supernatant was stored at −80°C until further experimentations. The protein concentration of the samples was determined following the method of Lowry using bovine serum albumin as a reference standard.
Measurement of prooxidant and antioxidant biomarkers
ROS production in brain homogenates of young and old control groups and rapamycin exposed groups was assessed using 2, 7-dichlorodihydrofluorescein diacetate dye following the published protocol with slight modifications. 22 The data of ROS generation in treated groups are expressed as percent change of control. The protein carbonyl (PCO) contents were analyzed by DNPH method as described earlier. 23 The values of PCO are given as nmol/mg protein. Moreover, the determination of lipid hydroperoxidation (LHP) was measured according to the FOX-2 method. 24 Briefly, brain homogenates (50 μL) were added to 950 μL of assay buffer (containing 100 μM xylenol orange, 250 μM ammonium ferrous sulfate, 90% methanol, 4 mM butylated hydroxytoluene, and 25 mM H2SO4) and incubated at room temperature for 30 minutes. Then, the absorbance was read at 560 nm after removal of any flocculated material by centrifugation. The signal was read against a H2O2 standard curve. The data of LHP are expressed as μM. The level of intracellular calcium ion in all the experimental groups was determined fluorometrically using Quin-2AM fluorescent dye following the method described earlier by us, 25 and the data are presented in nM. In addition, accumulation of nitrite in the supernatant, an indicator of the production of nitric oxide (NO), was determined by a colorimetric assay using Griess reagent (0.1% N-(1-naphthyl) ethylene diamine dihydrochloride, 1% sulfanilamide, and 5% phosphoric acid) following the protocol of Green et al. 26 The values of NO are given in μM/g protein. Reduced glutathione (GSH) level was determined by the standard method, 27 and the data are expressed as μM/mg protein. Furthermore, the activity of superoxide dismutase (SOD) was measured following the reported method 28 using NADH as a substrate, and the SOD activity is expressed as units per milligram protein. The catalase activity in brain homogenate of entire experimental group was measured by the H2O2 degradation assay following the earlier described protocol, 29 and the data are expressed as micromole H2O2 decomposed per minute per milligram protein.
Analysis of mitochondrial membrane potential
The mitochondrial membrane potential (MMP) was measured by flow cytometry using JC-1 dye that selectively targets mitochondria, and cells were isolated from the whole brain following the protocol of Singh et al., 30 with slight modifications. The brain of entire experimental group was finely sectioned in Hanks balanced salt solution and trypsinized at 37°C for 5 minutes using 0.01% trypsin/10 mM EDTA. Then, the brain tissue was disintegrated, dissociated, and finally passed through a nylon mesh (50 μm diameter) to obtain a single cell suspension. The cell suspension was further subjected to centrifugation at 700 g for 5 minutes, and the pellet thus obtained was resuspended in phosphate-buffered saline (PBS). The cells were incubated with JC-1 dye (10 μM) at 37°C for 15 minutes. Then, the cells were washed twice with PBS and finally resuspended in 0.3 mL PBS. Subsequently, the cells were analyzed with flow cytometer (FACSCalibur, BD Biosciences). The data were collected from at least 10,000 cells per event from three independent experiments.
Flow cytometric evaluation of apoptosis
The brain cells were isolated from the whole brain of all the experimental groups following the protocol of Singh et al., 30 and apoptotic cell death was assessed by flow cytometry using Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI). In brief, cells were resuspended in 500 μL of PBS and allowed to stain with 5 μL of Annexin V-FITC (Apoptosis Detection Kit; BD Pharmingen) and 5 μL of PI for 1 hour in the dark. Then, the apoptotic changes were quantified using flow cytometer (FACSCalibur; BD Biosciences). The data were collected from at least 10,000 cells per acquisition from three independent experiments.
Gene expression analysis by qRT-PCR
To study the effect of rapamycin on autophagy and consequently in brain aging, the expression of marker genes associated with autophagy (LC3, p62, and Beclin-1), aging (sirtuin-1, sirtuin-2, and Foxo3), neurodegeneration/neuroprotection (postsynaptic density-95, PSD95; myelin basic protein, MBP; and neuron specific enolase, NSE), and neuroinflammation (IL-1β, IL-6, and TNF-α) were studied in rat brain of all the experimental groups by qRT-PCR following the method as described earlier by us. 31 The alterations in gene expression are expressed in relative quantification. The primer sequences are available on request.
Protein expression analysis by western immunoblotting
The expression of proteins associated with autophagy (LC3, p62, and Beclin-1), aging (sirtuin-1, sirtuin-2, and Foxo3a), neurodegeneration/neuroprotection (PSD95, MBP, and NSE), and neuroinflammation (IL-1β, IL-6, and TNF-α) was studied in the experimental and control groups by western blotting following the protocol of our earlier publication. 31 Briefly, brain tissues were lysed with CelLytic™ Mammalian Tissue Lysis/Extraction Reagent in presence of 1X protease inhibitor cocktail. The amount of protein was determined by the method of Lowry, and equal amounts (50 μg) of protein were electrophoresed in 10%–12% SDS. Then, gels were transferred on PVDF membrane. After blocking, the membranes were incubated overnight at 4°C with primary antibodies specific for LC3 (1:1,000), p62 (1:1000), Beclin-1 (1:1000), PSD95 (1:1000), MBP (1:1000), NSE (1:1000), sirtuin-1 (1:1000), sirtuin-2 (1:1000), Foxo3a (1:500), IL1-β (1:1000), IL-6 (1:1000), TNF-α (1:1000), and β-actin (1:10,000). The membranes were then washed and incubated for 2 hours at room temperature with secondary anti-primary antibodies conjugated with horseradish peroxidase (Calbiochem). The blots that developed protein bands were quantified using SuperSignal West Femto Chemiluminescent Substrate™ (Thermo Fisher Scientific) and Bio-Rad VersaDoc™ Imaging System 4000 (Bio-Rad, PA). β-actin was used as endogenous control to normalize the data.
Preparation of synaptosomes
Before studying the effects of rapamycin-induced activation on autophagy on the activities of Na+/K+-ATPase, Ca2+-ATPase, and acetyl cholinesterase (AChE), synaptosomes from the brain homogenates of all the experimental groups were prepared following the Ficoll density gradient method of Torlińska et al. 32 The protein content in synaptosomes was determined following the method of Lowry.
Measurement of Na+/K+-ATPase activity
The Na+/K+-ATPase activity was determined by measuring the amount of inorganic phosphate (Pi) liberated from ATP during the incubation of synaptosomes prepared from brain homogenates of all the experimental groups. 33 The reaction mixture containing synaptosomes (50 μg protein) and assay buffer (100 mM NaCl, 20 mM KCl, 2 mM ATP-disodium salt, 30 mM Tris-HCl buffer, pH 7.4) in a final volume of 1.0 mL was preincubated at 37°C for 10 minutes to allow the reaction of ouabain (1.5 mM) with ATPase. The reaction was initiated by the addition of ATP and terminated after 15 minutes of incubation by the addition of 500 μL of 15% (w/v) trichloroacetic acid (TCA). The amount of Pi liberated was estimated according to the method of Fiske and Subbarow. 34
Measurement of Ca2+-ATPase activity
The Ca2+-ATPase activity was assayed following the established method. 35 Briefly, the reaction mixture (1.0 mL) containing 50 mM imidazole-HCl buffer (pH 7.5), 0.4 mM CaCl2, 2 mM ATP, and the synaptosomes (50 μg protein) were suspended in 0.32 M sucrose. After 15 minutes of incubation at 37°C, the reaction was stopped by adding 0.5 mL of ice-cold 10% TCA. The amount of Pi liberated was estimated according to the method of Fiske and Subbarow. 34
Measurement of AChE activity
AChE activity was determined following the method described earlier. 36 Briefly, the reaction mixtures (2.0 mL) containing brain homogenates, 50 mM Tris-HCl (pH 8.0), 240 mM sucrose, and 120 mM NaCl were added to 0.03 mL of 0.5 mM 5.5%-DTNB and 0.05 mL of 10 mM acetylthiocholine iodide as a substrate to start the reaction. The progress of reaction was followed using UV/VIS spectrophotometer by the increase in absorbance (ΔOD) at 412 nm.
Statistical analysis
The experimental data are expressed as mean ± SEM of three independent experiments. The statistical significance of differences among various groups was calculated by one-way analysis of variance (ANOVA) followed by Bonferroni's post hoc test, and intragroup variation between control and treatment group was performed by Student's t-test. Statistical analysis was performed using GraphPad Prism 5 version 5.01 software package (San Diego, CA). In all analyses, a probability value of p < 0.05 was considered as statistically significant. The symbol * indicates significant difference compared with young control group, and symbol # indicates significant difference compared with old control group.
Results
Rapamycin alleviates age-associated oxidative stress
To evaluate the effect rapamycin-induced activation of autophagy had on age-associated impairment of redox homeostasis, several prooxidant biomarkers such as ROS, PCO, LHP, intracellular calcium ion [Ca2+]i, and nitric oxide (NO), and anti-oxidant biomarkers such as reduced GSH, SOD and catalase were measured in young and old aged rats (Fig. 1). The level of prooxidants such as ROS (161.5% ± 15.5%), PCO (4.18 ± 0.52 nmol/mg protein), LHP (25.3 ± 2.1 μM), [Ca2+]i (489.4 ± 27.4 nM), and NO (0.74 ± 0.06 μM/g protein) was found significantly (p < 0.05) increased in old aged rats compared to young aged control rats. Rapamycin supplementation in old aged rats significantly protected against age-related alterations in prooxidants by reversing the levels of ROS (126.3% ± 12.3%), PCO (3.15 ± 0.28 nmol/mg protein), LHP (21.6 ± 1.9 μM), [Ca2+]i (341.4 ± 21.3 nM), and NO (0.62 ± 0.04 μM/g protein), closer to the levels of young control rats. Conversely, the exposure of rapamycin did not alter the levels of prooxidants in young rats (Fig. 1A–E). In contrast, a significant reduction in the level of antioxidants such as GSH (18.5 ± 1.7 μM/mg protein), SOD (6.4 ± 0.8 U/mg protein), and catalase (7.5 ± 0.7 μmol H2O2 decomposed/min/mg protein) was also observed in old aged rats compared to young control. However, the exposure of rapamycin significantly elevated the levels of GSH (21.8 ± 2.6 μM/mg protein), SOD (16.4 ± 1.3 U/mg protein), and catalase (11.3 ± 1.2 μmol H2O2 decomposed/min/mg protein) in old aged rats. Furthermore, the rapamycin did not show a significant response on the levels of antioxidants in young rats (Fig. 1F–H). Taken together, the data revealed that rapamycin-induced activation of autophagy significantly protected the brain against aging-dependent impairment of redox homeostasis.

Rapamycin mediates protection against age-dependent impairment of redox status. Effect of rapamycin supplementation on
Effect of rapamycin-induced autophagy, activation of MMP, and apoptosis in aging rat brain
MMP is an important indicator for initiation of apoptotic cell death. Moreover, the reduction in MMP is also associated with loss of permeability of the mitochondrial membrane. The MMP in rat brain cells was determined by flow cytometry using JC-1 dye (Fig. 2A). A significant induction of mitochondrial dysfunction was observed during aging process as revealed by depolarization of MMP (57.12%) in old aged rat brain cells compared to young control group (15.02%). Rapamycin supplementation improved the age-associated loss of MMP by reducing the depolarization of MMP (23.46%). However, the exposure of rapamycin did not show any significant response on MMP (9.01%) in brain cells of young rats. Thus, our data confirmed the age-related mitochondrial dysfunction and also its protection with rapamycin supplementation. Next, the percentage of apoptotic cell death in brain cells was determined using Annexin V-FITC Apoptosis Detection Kit, based on its affinity toward mitochondrial phosphatidylserine on the membrane (Fig. 2B). The data demonstrated the increased percentage of early apoptotic cells (12.81%) and late apoptotic cells (12.26%) in brain cells during aging of rats. Furthermore, rapamycin supplementation reversed the age-related induction in apoptotic cell death by decreasing the percentage of cells (7.17%) in late phase of apoptosis in old aged rats. However, rapamycin supplementation did not change the population of brain cells of young rats in late phase of apoptosis. Taken together, the results confirmed the protection of mitochondria with rapamycin.
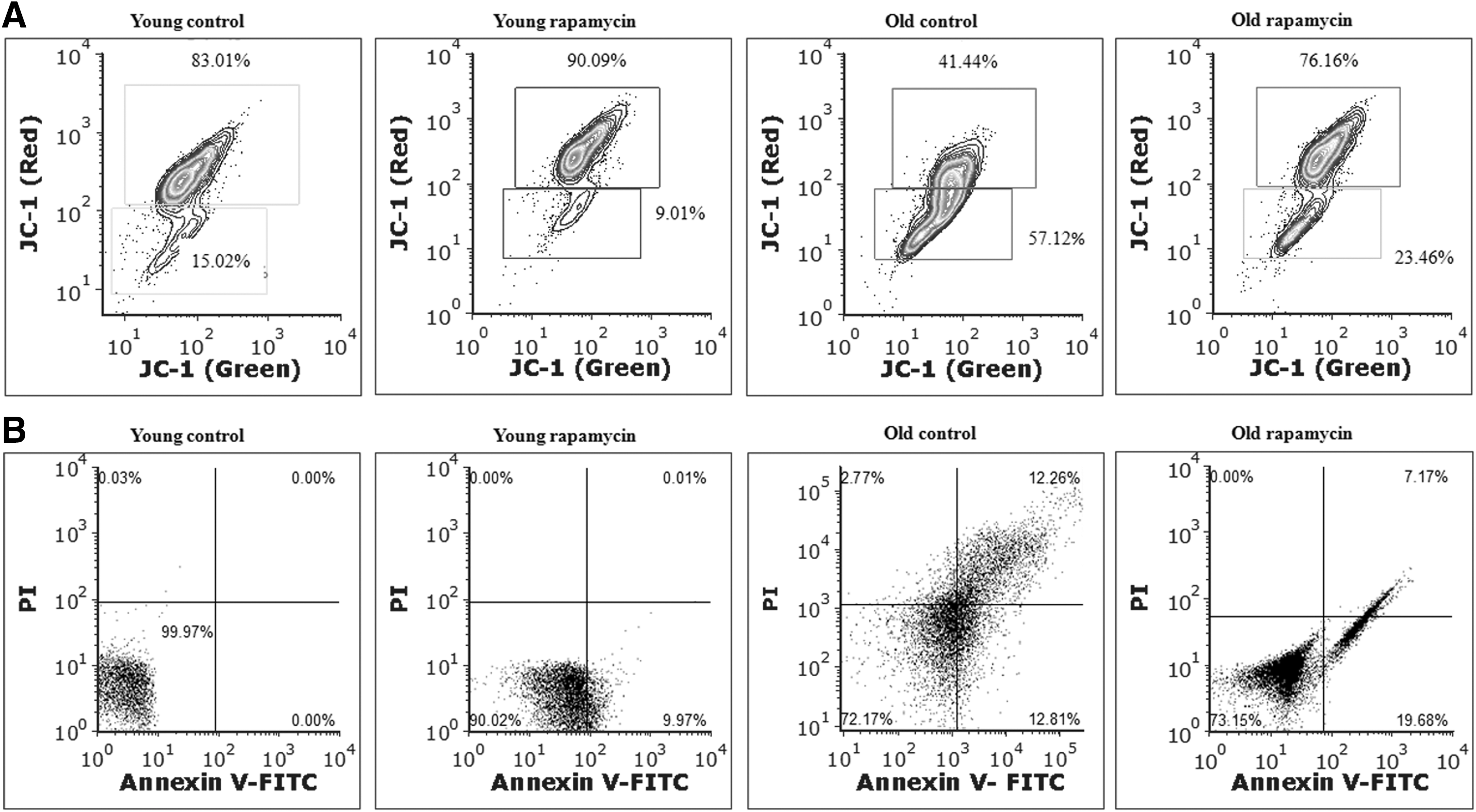
Rapamycin mediated protection of aging rat brain against mitochondrial membrane depolarization and apoptotic cell death.
Rapamycin-induced activation of autophagy in aging rat brain
We also evaluated the age-dependent level of autophagy, as well as rapamycin-induced activation of autophagy in young and old rat brain, by measuring the expression of autophagy markers during aging. qRT-PCR results revealed the age-dependent decrease in the mRNA expression of autophagy markers such as LC3 (0.77 ± 0.06-fold), Beclin-1 (0.53 ± 0.05-fold), and p62 (0.43 ± 0.05-fold) compared to normal young rats. The data also showed that rapamycin supplementation significantly (p < 0.05) induced the expression of LC3, Beclin, and p62 to 2.5 ± 0.2, 1.8 ± 0.2, and 1.6 ± 0.1-fold in young aged rats and 1.7 ± 0.12, 0.95 ± 0.09, and 0.85 ± 0.07-fold in old aged rats compared to normal young aged rats (Fig. 3A). Western blot data further confirmed the age-dependent downregulation of autophagy-related marker proteins and suppression of autophagy process, as well as induction of aging-associated defective autophagy in old aged rat brain following the exposure of rapamycin (Fig. 3B, C). The expression of p62 protein was found at significantly higher level in old rats in comparison to p62 mRNA expression. This unequal expression between protein and mRNA might be due to shorter half-life and turnover of mRNA.
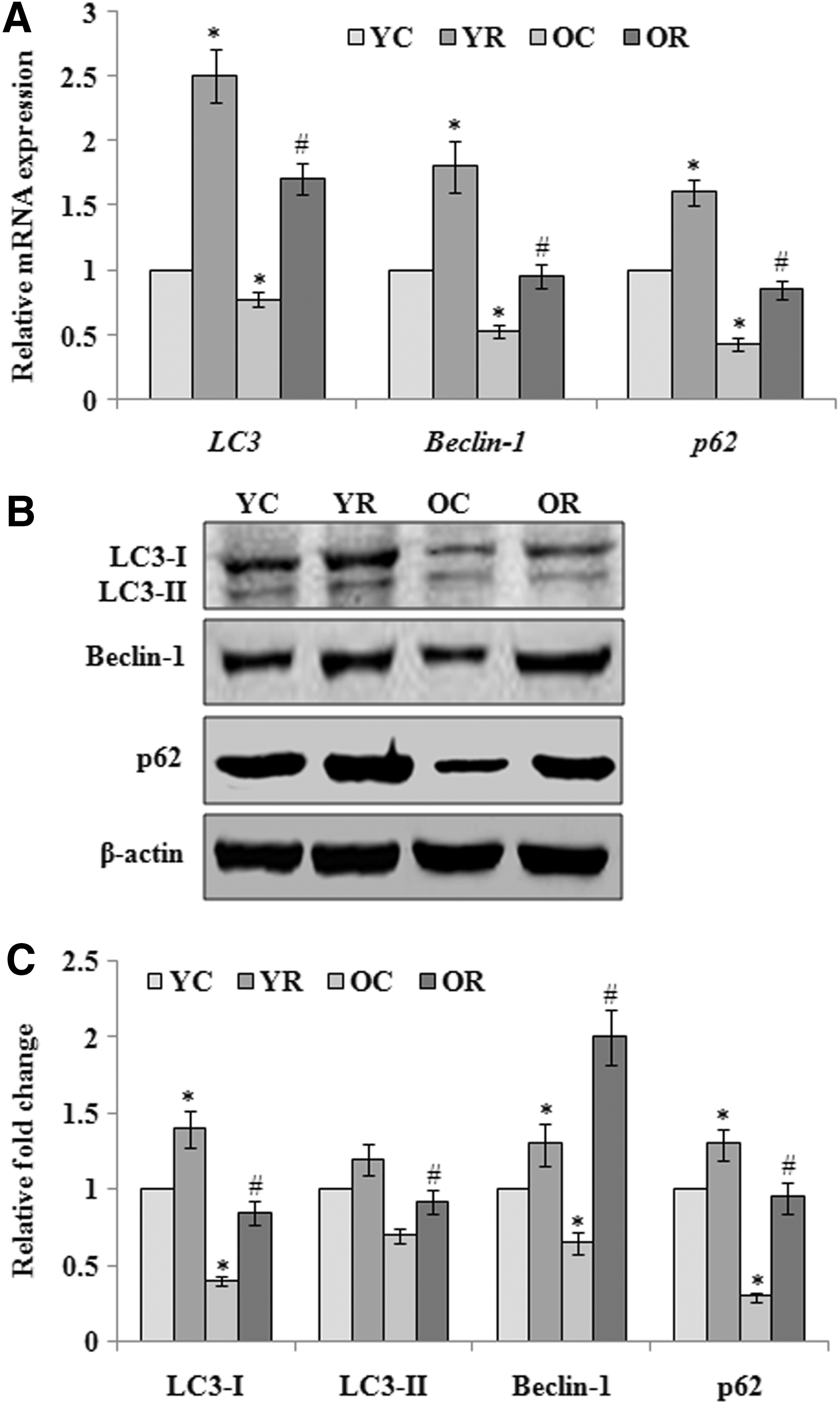
Rapamycin induced activation of autophagy and expression (mRNA/protein) analysis of autophagy markers.
Effect of rapamycin-induced activation of autophagy on expression of markers associated with aging, neuroinflammation, and neurodegeneration
Next, we investigated the consequences of autophagy activation on the aging-associated changes in markers of aging (sirtuin-1, sirtuin-2, and Foxo3A), neuroinflammation (IL-1β, IL-6, and TNF-α), and neurodegeneration (PSD95, MBP, and NSE) transcriptional and translational level (Fig. 4A–C). qRT-PCR and western blot data revealed that the expression of sirt-1 and Foxo3a was significantly decreased in old aged rats, whereas sirt-2 was increased that were reversed upon rapamycin-induced activation of autophagy. The levels of neuronal markers such as PSD95, MBP, and NSE were also found decreased with advancing of age. Rapamycin-induced activation of autophagy was observed to protect the brain during aging by reversing the levels of these neuronal markers closer to the normal young rats. Furthermore, neuroinflammatory response was also found significantly activated in old aged rats as evidenced by the elevated levels of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α in the brain. However, activated autophagy was observed to reduce the levels of these pro-inflammatory cytokines in the brain of old aged rats and, thus, could minimize the inflammatory response. Taken together, the data corroborate that the activation of autophagy in old age played significant role in neuroprotection of aging brain.
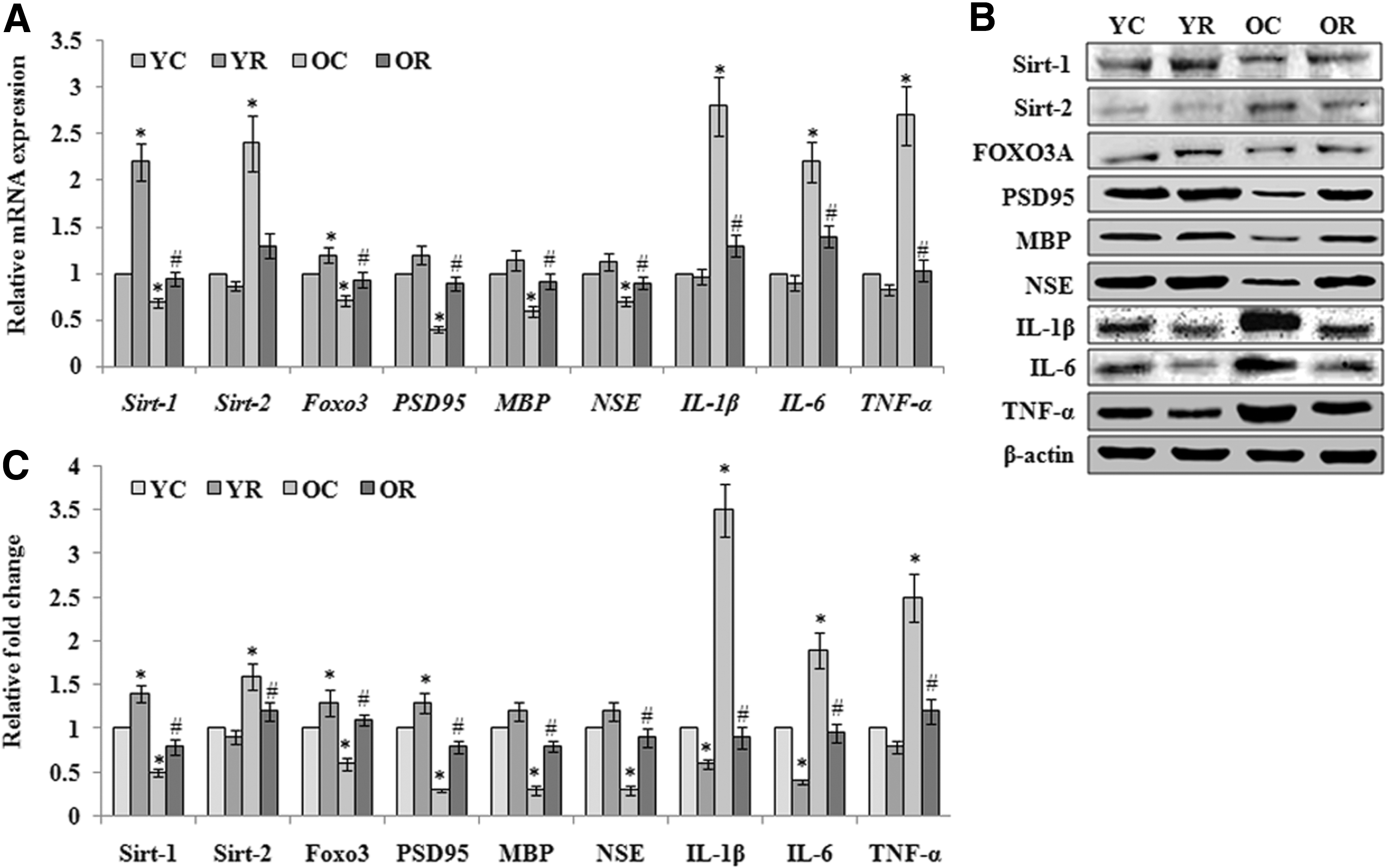
Rapamycin-induced activation of autophagy provides neuroprotection in aging rats.
Effect of rapamycin-induced activation of autophagy on membrane transporters and AChE activity
To determine the effect of rapamycin-induced activation of autophagy on brain cell membrane, the activities of transporters such as Na+/K+-ATPase, Ca2+-ATPase, and enzyme AChE were carried out in synaptosomal preparation and highlighted in Figure 5A–C, respectively. The results demonstrated that the activities of Na+/K+-ATPase (130.5 ± 15.9 nmol Pi/mg protein/hour), Ca2+-ATPase (397 ± 30 nmol Pi/mg protein/hour), and AChE (72.2% ± 6.8%) were significantly (p < 0.05) decreased with the progression of age compared to normal young control. Furthermore, rapamycin supplementation significantly reversed the age-related impairment of activities of Na+/K+-ATPase (194.4 ± 20.6 nmol Pi/mg protein/hour), Ca2+-ATPase (590 ± 52 nmol Pi/mg protein/hour), and AChE (84.9% ± 8.8%). However, nonsignificant changes in these parameters were observed following the exposure of rapamycin in young rats.
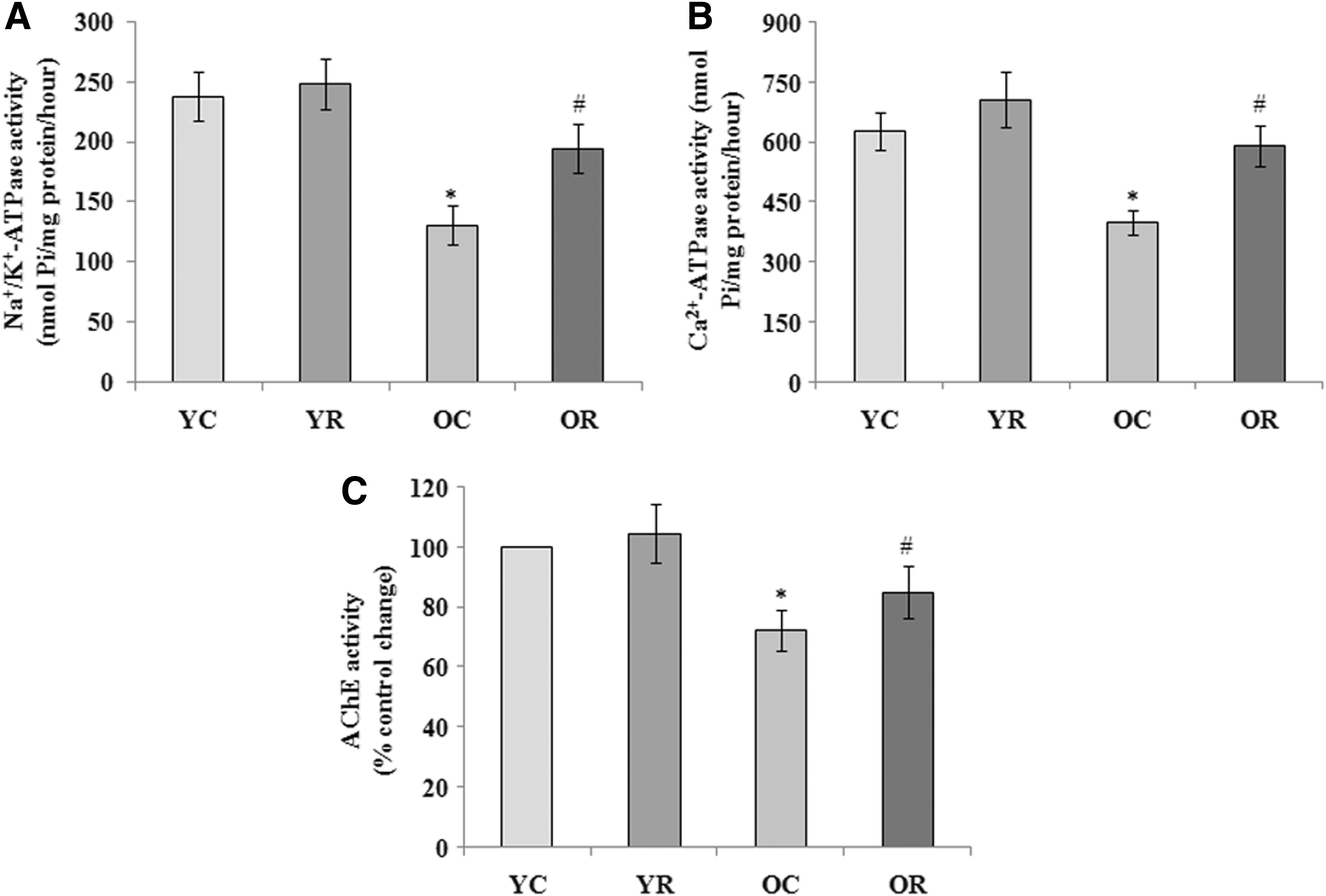
Effect of rapamycin-induced activation of autophagy on
Discussion
The ROS-mediated oxidative damage to macromolecules is a probable culprit in cellular senescence and aging. 37 Herein, we report the aging-dependent induction in level of prooxidant biomarkers and decrease in level of antioxidant biomarkers in brain tissue of old aged rats that were reversed upon exposure to rapamycin. An accumulation of oxidatively damaged proteins with advancing of age increases the risk for age-related disorders, including neurological diseases. 38 In addition, a defective proteolytic activity has been observed during aging that further contributes to the accumulation of damaged cellular components. 39
Aging brain is also associated with a compromised antioxidant defense mechanism that increases the vulnerability of the brain toward oxidative damage. 40 Nitric oxide (NO) is synthesized upon activation of N-methyl-D-aspartate glutamate receptors in the nervous system that further induces calcium ion (Ca2+) influx and prolonged ROS generation. 41 However, the increased level of NO and ROS have been shown to disrupt autophagic functions. 42 The Ca2+ influx mediated activation of caspases and calpains has been implicated in reduction of neuronal autophagy. 43 In line, we have shown the neuroprotective activity of rapamycin against oxidative stress in the brain. 31
The oxidative damage to mitochondria leads to depletion of ATP, Ca2+ influx, loss of MMP, and apoptosis. The age-dependent loss of MMP increases the membrane permeability that facilitates the release of the cytochrome c protein in cytoplasm to initiate apoptosis. 44 The apoptosis pathways have been clearly shown to be involved in age-related neurodegenerative disorders. 45 Similarly, we also observed the notable loss of MMP and induction of apoptosis in brain cells of aged rats. Moreover, the decreased activity of SOD has been reported to induce the loss of MMP and to activate the caspase cascade mediated apoptotic cell death. 46
Next, we have shown an age-dependent defect in the autophagy process in rat brain, evidenced by elevated expression of autophagy-related proteins such as LC3-II (microtubule-associated protein light chain 3), p62 (sequestosome-1), and Beclin-1. These autophagy-related proteins help in regulation and formation of autophagosomes. 47,48 It has been well documented that the basal level of autophagy continuously eliminates damaged cell organelles and protein aggregates and, thus, contributes to cellular homeostasis and functional integrity. 49 An age-dependent decline in autophagy process in brain and other tissue contributes to different aspects of the aging phenotype and age-related diseases. 21,50 The elevated expression of autophagy-related defective genes has been proven effective in improving life span and providing resistance to oxidative stress. 51 Komatsu et al., 52 have shown that the conditional knockout of ATG7 in mice causes premature death and shortened life span. In contrast, overexpression of ATG8a protein in neurons has shown dramatic increase in life span of Drosophila that also enhances the removal of damaged proteins. 51 Earlier, we have also shown the rapamycin-induced activation of autophagy and PI3K/Akt1/CREB pro-survival pathways through inhibition of mTOR that provided neuroprotection against amyloid-β-induced synaptic neurotransmission dysfunction and neurodegeneration in adult rats. 31
The activation of the beclin-1 class III PI3K dependent autophagy, instead of mTOR dependent autophagy, may also be a beneficial approach to promote the mammalian health span and life span. 53 In our study, the level of p62 significantly decreased in the brain tissue of control old rats which was further increased upon the treatment of rapamycin. Moreover, prolonged starvation leads to the restoration of SQSTM1/p62 expression level. This restoration of SQSTM1/p62 is completely mTORC1 independent and is determined by the transcriptional regulation and the availability of amino acids recycled by autophagy. 54
Sirtuin-1 (Sirt-1), NAD-dependent class III histone deacetylase enzyme, is localized predominantly in the nucleus of neurons and mainly regulates apoptotic mediators such as p53. 55 An age-dependent decrease in Sirt1 protein level has been observed in different cells and tissues that often lead to senescence, loss of dendrites, and neurotransmitter synthesis. 56 In addition, a significant decrease in Sirt-1 expression was also reported in older cells. 57 However an increased level of Sirt-1, through caloric restriction, has been shown to prolong the life span. 58 Sirt-1 has also been proposed to play a major role in neuroprotection that is thought to be mediated, in part, by the deacetylation of p53 and inhibition of apoptosis. Similarly, our results show increased expression of Sirt-1 after rapamycin treatment in aged rats, thus providing a mechanism to explain neuroprotection. Furthermore, activation of Sirt-1 has been shown to reduce oxidative stress and suppress the expression of pro-inflammatory cytokines. 59 However, Sirt-2 overexpression was shown to reduce the survival of healthy neurons. 60 Our study demonstrates that rapamycin downregulates the expression of Sirt-2, suggesting its antiapoptotic effect. To further investigate the effect of Sirt-2 function during aging, we examined the levels of FOXO3A protein level in the aging brain. FOXO is a pro-longevity factor that plays an important role in degradation of accumulated protein aggregates to maintain the protein homeostasis through regulation of autophagy and the ubiquitin-proteasome system. 61 Sirt-1 binds and deacetylates p53 and FOXO transcription factors for controlling their activity. Moreover, FOXO proteins have been reported to detoxify the ROS accumulation by upregulating the free radical scavenging enzymes, including SOD and catalase. 62 In line, our study also demonstrated the reduced level of FOXO3A in the brain tissue of old rats that was reversed to the normal level upon supplementation of rapamycin.
Subsequently, we evaluated the effect of autophagy on neuroinflammation and neurodegeneration in aging rats. Earlier, we have reported the elevated neuroinflammatory response in rat brain during aging that was significantly depressed by the treatment of metformin. 21 In rodents and humans, brain aging is associated with increased incidence of activated astrocytes and microglia. 63 Once activated, microglia produce several pro-inflammatory mediators, including cytokines such as IL-1β, IL-6, and TNF-a, cytotoxic complement molecules, ROS, and nitric oxide. 8 In addition, autophagy has been shown to play an essential function in controlling infectious and inflammatory diseases and immunity. 64 Previous reports have revealed that the autophagy inducer rapamycin recovers cognition ability through inhibition of amyloid-β and tau protein accumulation and also interferes with several signaling cascades 31,65 that include interactions between oxidative stress and neuroinflammation. 66 In line with these observations, we also noted the decreased level of pro-inflammatory cytokines after the activation of autophagy during brain aging in rats. Moreover, rapamycin treatment demonstrated possible neuroprotection through the reduction of microglial activation and induction of autophagy process in an experimental traumatic brain injury model. 67 Furthermore, the beneficial effect of rapamycin on the aging brain might be due to the fact that it triggers a nutrient signal to the hypothalamus which generates an anorexic response to reduce food consumption. 68,69 This anorexic response is also responsible for decreased body weight and fat mass along with increased lean muscle mass and high physical activity in older animals. 70
An aging brain has been associated with decrease in synaptic connectivity, 71 and a growing body of evidence reveals that autophagy is capable of regulating synaptic function at presynaptic and postsynaptic terminals. 72 In addition, autophagy has also been shown to regulate the synaptic growth and plasticity, neurotransmission, and cognitive ability. 31,73 Similarly, we also noticed the autophagy-mediated maintenance of neuronal markers such as PSD95, MBP, and NSE at normal level in the brain of old aged rats. The synaptic protein PSD95 has been suggested to be involved in aging and age-dependent neurological diseases. 74 Xing et al. 75 have suggested that the decreased MBP and degeneration of myelin with advancing of age play a critical role in the loss of spiral ganglion neurons and the subsequent decline of the auditory nerve function during age-related hearing loss. Moreover, age-related changes in the levels of NSE and MBP have also been reported in cerebrospinal fluid. 76
Membrane bound enzymes play vital roles in maintenance of ionic homeostasis. An altered expression of membrane bound transporters has been shown to be associated with many chronic diseases, including neurological diseases and aging. 77 In the present investigation, the activity of Na+/K+-ATPase, responsible for the regulation of intracellular pH, cell volume, and action potential in neurons, was found to be decreased in synaptosomes of aged rat brain. Our findings corroborate the earlier studies showing decreased activity of membrane bound transporters during aging. 78 The age-dependent decreased Na+/K+-ATPase and Ca2+-ATPase activity in brain has been attributed to the progressive production of ROS which leads to lipid peroxidation and damage to the neuronal membrane. An elevation in levels of intracellular Na+ and Ca2+ may impair proteasome function and autophagy, 79 which may result in dysregulated cellular homeostasis. 80 Moreover, Na+/K+-ATPase pump also mediates autophagy dependent cell death known as autosis. The prolonged activation of cellular autophagy at high level due to starvation, hypoxia, or treatment with autophagy inducing peptides triggers autosis. 81 The cholinergic neurotransmission disruption has also been shown in old age. 82 Moreover, the oxidative stress mediated decrease in AChE activity has been observed in old aged rat brain. 21 In the present investigation, we report autophagy mediated reversal of aging-induced reduced AChE activity.
Conclusion
Our study demonstrates that rapamycin induced activation of autophagy provides possible neuroprotection against aging-associated oxidative stress, apoptotic cell death, neuroinflammation, and neurodegeneration in rat brain. The findings also suggest that autophagy inducers may be suitable therapeutic interventions for slowing down brain aging and age-related neurological disorders.
Footnotes
Acknowledgments
A. K. Singh acknowledges Dr. D. S. Kothari Post Doctoral Fellowship scheme of University Grant Commission, New Delhi, India for providing financial support (F.4-2/2006(BSR)/BL/14-15/0326) and fellowship. Department of Biochemistry is supported by a FIST grant of DST-SERB, Government of India and DRS-SAP grant of University Grants Commission, New Delhi.
Author Disclosure Statement
The authors declare that they have no conflicts of interest.